一种La2Zr2O7上转换发光材料及其制备方法与流程
本发明涉及一种上转换发光材料,尤其涉及一种la2zr2o7上转换发光材料及其制备方法,属于稀土材料技术领域。
背景技术:
la2zr2o7为a2b2o7型结构晶体,其具有声子能量较低的特性使其作为上转换发光材料基质可在较低能量激发源下激发发光,并且其具有优秀的化学稳定性及热稳定,使其有望成为优质的上转换发光材料基质。将er3+作为激活剂而yb3+作为敏化剂共掺杂进入la2zr2o7晶格,将可观察到在yb3+敏化剂作用下增强的er3+的上转换发光现象。目前,la2zr2o7主要采用高温固相合成法、共沉淀法、溶胶凝胶法进行制备,采用高温固相法合成温度较高(1550℃-1650℃),且所制备产物存在着颗粒度偏大、产物纯度不高、产物需进行球磨而增加颗粒表面缺陷、本体晶格缺陷难控制和难于进行均匀掺杂等缺点从而降低其发光材料的性能。
技术实现要素:
本发明要解决的技术问题是:提供一种la2zr2o7上转换发光材料及其制备方法,该方法形成的凝胶在较低温度下烧结即可得到分散均匀的粉体材料,有效的解决了上述存在的问题。
本发明的技术方案为:一种la2zr2o7上转换发光材料,所述材料的化学通式为la2-2x-2yzr2o7:er2x,yb2y,其中:x为er离子取代la离子的摩尔数之比,x的取值范围为0<x≤0.3,y为yb离子取代la离子的摩尔数之比,y的取值范围为0<y≤0.5,冒号“:”表示为tb和m离子共同掺杂,优选x的取值范围为0.01≤x≤0.1,y的取值范围为0.1≤y≤0.3。
一种la2zr2o7上转换发光材料的制备方法,所述方法包括以下步骤:
一、分别配制稀土盐溶液;
二、按化学通式la2-2x-2yzr2o7:er2x,yb2y中各la、zr、er和yb元素的摩尔比,分别移取步骤一中的相应量的la、zr、er和yb溶液混合均匀,得到混合金属离子溶液;
三、按质量比分别称量丙烯酰胺与n,n-亚甲基双丙烯酰胺,加入到步骤二制备的混合金属离子溶液中,混合均匀;
四、将步骤三所得混合均匀溶液置于50~90℃的水浴锅中水浴搅拌,按反应体系溶液的体积比加入过硫酸盐溶液,水浴反应0.5~6h形成湿凝胶;
五、将步骤四所得的湿凝胶置于60~120℃的干燥箱中烘干5~24h,获得干凝胶;
六、将步骤五所得的干凝胶转移至坩埚内,将坩埚置于炉中700~1200℃煅烧3-24h,随炉冷却至室温,研磨,即得到化学通式为la2-2x-2yzr2o7:er2x,yb2y上转换发光材料。
所述步骤一中,配制稀土盐溶液的方法为分别将氧化镧、氧化铒、氧化镱、氧化锆溶解在浓硝酸中。
所述步骤一中,配制稀土盐溶液的方法为采用可溶性稀土盐及锆盐分别配制含稀土溶液和含锆溶液;
所述可溶性稀土盐分别为含la、er和yb对应的硝酸盐、乙酸盐、盐酸盐或硫酸盐等可溶性稀土盐中的一种或几种,优选为硝酸盐。
所述含锆溶液为硝酸锆、盐酸锆、硫酸锆、乙酸锆、草酸锆、磷酸锆、碳酸锆、硝酸氧锆、碳酸氧锆或氯氧化锆等可溶性含锆化合物中的一种或几种,优选为硝酸氧锆和氯氧化锆中的一种或两种。
所述稀土溶液和含锆溶液的浓度为0.01~5mol/l,优选稀土溶液和含锆溶液的浓度为0.1~1mol/l。
所述步骤二的混合金属离子溶液中la、er和yb总离子浓度为0.01-3mol/l,优选混合金属离子溶液中la、er和yb总离子浓度为0.1-1mol/l。
所述步骤三中的丙烯酰胺与n,n-亚甲基双丙烯酰胺的质量比为2~10:1,优选丙烯酰胺与n,n-亚甲基双丙烯酰胺的预设质量比为4~7:1。
所述步骤四中的过硫酸盐溶液的质量分数为3~30%(优选为10~15%),加入体积为反应体系溶液体积的0.01~10%(优选为1~5%),所述过硫酸盐为过硫酸铵、过硫酸钾或过硫酸钠中的一种或几种,优选为过硫酸铵和过硫酸钠中的一种或两种。
本发明的有益效果是:与现有技术相比,采用本发明的技术方案,高分子网络凝胶法利用高分子凝胶形成网络的阻碍作用,使粒子在溶液中的移动受到限制,接触和团聚机会大大减小,粒子可以均匀地分散在网络中,从而有利于生成平均粒径小、分散均匀、团聚少的粉体材料;且该方法操作简单,对原料要求简单,原料为无机盐水溶液即可;凝胶法的制备可以在酸性、中性、碱性条件下制得;该方法形成的凝胶在较低温度下烧结即可得到分散均匀的粉体材料。
干凝胶经700℃煅烧即可制备纯相er3+、yb3+共掺杂la2zr2o7晶体,煅烧温度,无杂相,结晶度高,煅烧温度较传统固相合成法降低了500℃以上。本发明所需原材料均可从市场公开购买,使用设备为普通设备无特殊要求,且制备工艺简单,成本低,易于放大投入批量生产,取得了很好的使用效果。
附图说明
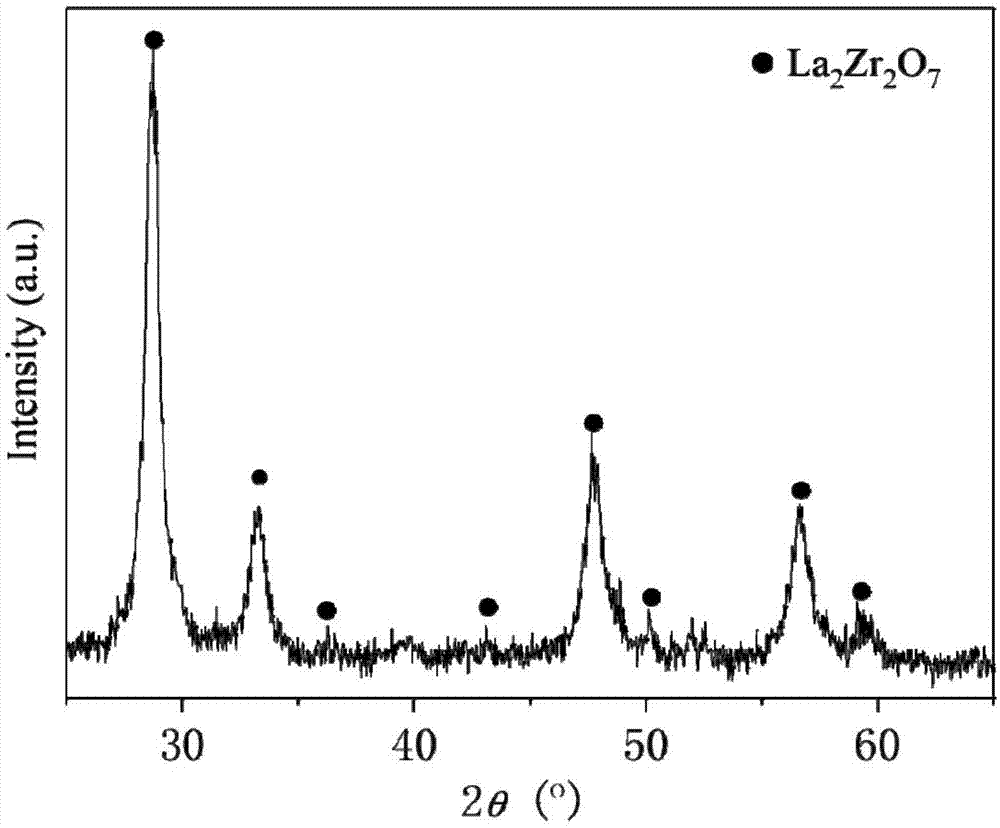
图1为本发明具体实施例1制备的la1.7zr2o7:er0.1,yb0.2上转换发光材料x射线粉末衍射图;
图2为本发明具体实施例2制备的la1.86-2yzr2o7:er0.14,yb2y(y=0.16,0.20,0.24)系列上转换发光材料在980nm光激发下的室温发射光谱图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将参照本说明书附图对本发明作进一步的详细描述。
实施例1:高分子网络凝胶法制备la1.7zr2o7:er0.1,yb0.2上转换发光材料:以la(no3)3·6h2o、er(no3)3·6h2o、yb(no3)3·6h2o和zro(no3)2·2h2o为主要原料,配制含la、er、yb和zr溶液的浓度分别为1.0mol/l、0.2mol/l、0.2mol/l、1.0mol/l的溶液。分别取浓度为1.0mol/l的la3+溶液42.50ml、0.2mol/l的er3+溶液12.50ml、0.2mol/l的yb3+溶液25.00ml、1.0mol/l的zr4+溶液50.00ml和100.00mlh2o至500ml烧杯中混合均匀获得混合离子溶液;按质量比为6:1分别称量丙烯酰胺12g与n,n-亚甲基双丙烯酰胺2g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于80oc水浴锅中水浴搅拌0.5h,加入新制质量分数为20%的过硫酸铵溶液1.00ml,继续水浴搅拌3h形成湿凝胶;将湿凝胶置于100oc烘箱中干燥12h获干凝胶;将干凝胶转移至坩埚中于850oc的马弗炉中煅烧10h,随炉冷却至室温、研磨,即获得la1.7zr2o7:er0.1,yb0.2上转换发光材料,样品的x射线粉末衍射图见附图1所示。
实施例2:高分子网络凝胶法制备la1.86-2yzr2o7:er0.14,yb2y(y=0.16,0.20,0.24)系列上转换发光材料:以lacl3·6h2o、ercl3·6h2o、ybcl3·6h2o和zrocl2·8h2o为主要原料,配制含la、er、yb和zr溶液的浓度分别为0.5mol/l、0.1mol/l、0.2mol/l、1.0mol/l的溶液。分别取三份浓度为0.1mol/l的er3+溶液28.00ml和1.0mol/l的zr4+溶液40.00ml至装有100mlh2o的500ml烧杯a、烧杯b和烧杯c中,按照yb3+掺杂量设计为16%、20%和24%,在烧杯a、烧杯b和烧杯c中分别加入0.5mol/l的la3+溶液61.6ml、58.4ml和55.2ml,随后在烧杯a、烧杯b和烧杯c分别加入0.2mol/l的yb3+溶液32.00ml、40ml和48ml,搅拌均匀获得混合离子溶液;按质量比为5:1分别称量丙烯酰胺10g与n,n-亚甲基双丙烯酰胺2g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于70oc水浴锅中水浴搅拌1h,加入新制质量分数为10%的过硫酸钠溶液3.00ml,继续水浴搅拌5h形成湿凝胶;将湿凝胶置于120oc烘箱中干燥6h获干凝胶;将干凝胶转移至坩埚中于900oc的马弗炉中煅烧12h,随炉冷却至室温、研磨,即获得la1.86-2yzr2o7:er0.14,yb2y(y=0.16,0.20,0.24)系列上转换发光材料。
实施例3:高分子网络凝胶法制备la1.7zr2o7:er0.1,yb0.2上转换发光材料:以la2o3、er2o3、yb2o3和zrocl2·8h2o为主要原料,称取一定量的稀土氧化物溶解在浓硝酸中,经加热分解过量的浓硝酸后获得相应含稀土溶液,分别配制含la、er、yb和zr溶液的浓度分别为1.0mol/l、0.2mol/l、0.2mol/l、1.0mol/l的溶液。分别取浓度为1.0mol/l的la3+溶液42.50ml、0.2mol/l的er3+溶液12.50ml、0.2mol/l的yb3+溶液25.00ml、1.0mol/l的zr4+溶液50.00ml和100.00mlh2o至500ml烧杯中混合均匀获得混合离子溶液;按质量比为6:1分别称量丙烯酰胺12g与n,n-亚甲基双丙烯酰胺2g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于80oc水浴锅中水浴搅拌0.5h,加入新制质量分数为20%的过硫酸铵溶液1.00ml,继续水浴搅拌3h形成湿凝胶;将湿凝胶置于100oc烘箱中干燥12h获干凝胶;将干凝胶转移至坩埚中于850oc的马弗炉中煅烧10h,随炉冷却至室温、研磨,即获得la1.7zr2o7:er0.1,yb0.2上转换发光材料,样品的x射线粉末衍射图见附图1所示。
实施例4:高分子网络凝胶法制备la1.7zr2o7:er0.1,yb0.2上转换发光材料:以la(no3)3·6h2o、er(no3)3·6h2o、yb(no3)3·6h2o和zr(no3)4·5h2o为主要原料,配制含la、er、yb和zr溶液的浓度分别为1.0mol/l、0.2mol/l、0.2mol/l、1.0mol/l的溶液。分别取浓度为1.0mol/l的la3+溶液42.50ml、0.2mol/l的er3+溶液12.50ml、0.2mol/l的yb3+溶液25.00ml、1.0mol/l的zr4+溶液50.00ml和100.00mlh2o至500ml烧杯中混合均匀获得混合离子溶液;按质量比为4:1分别称量丙烯酰胺6.0g与n,n-亚甲基双丙烯酰胺1.5g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于80oc水浴锅中水浴搅拌0.5h,加入新制质量分数为20%的过硫酸铵溶液1.00ml,继续水浴搅拌3h形成湿凝胶;将湿凝胶置于100oc烘箱中干燥12h获干凝胶;将干凝胶转移至坩埚中于850oc的马弗炉中煅烧10h,随炉冷却至室温、研磨,即获得la1.7zr2o7:er0.1,yb0.2上转换发光材料。
实施例5:高分子网络凝胶法制备la0.98zr2o7:er0.02,yb1.0上转换发光材料:以la2o3、er2o3、yb2o3和zr(so4)2·4h2o为主要原料,称取一定量的稀土氧化物溶解在浓硝酸中,经加热分解过量的浓硝酸后获得相应含稀土溶液,分别配制含la、er、yb和zr溶液的浓度分别为5.0mol/l、1.0mol/l、5.0mol/l、5.0mol/l的溶液。移液管分别移取浓度为5.0mol/l的la3+溶液29.80ml、1.00mol/l的er3+溶液3.00ml、5.0mol/l的yb3+溶液30.00ml、5.0mol/l的zr4+溶液60.00ml至250ml烧杯中混合均匀获得混合离子溶液;按质量比为2:1分别称量丙烯酰胺10g与n,n-亚甲基双丙烯酰胺5g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于90oc水浴锅中水浴搅拌0.5h,加入新制质量分数为3%的过硫酸铵溶液12.00ml,继续水浴搅拌0.5h形成湿凝胶;将湿凝胶置于60oc烘箱中干燥24h获干凝胶;将干凝胶转移至坩埚中于1200oc的马弗炉中煅烧3h,随炉冷却至室温、研磨,即获得la0.98zr2o7:er0.02,yb1.0上转换发光材料。
实施例6:高分子网络凝胶法制备la1.38zr2o7:er0.6,yb0.02上转换发光材料:以lacl3、er2(so4)3·8h2o、yb(ch3coo)3·4h2o和zr(ch3coo)4为主要原料,配制含la、er、yb和zr溶液的浓度分别为0.1mol/l、0.05mol/l、0.01mol/l、0.1mol/l的溶液。移液管分别移取浓度为0.1mol/l的la3+溶液13.80ml、0.05mol/l的er3+溶液12.00ml、0.01mol/l的yb3+溶液2.00ml、0.1mol/l的zr4+溶液20.00ml和152mlh2o至500ml烧杯中混合均匀获得混合离子溶液;按质量比为10:1分别称量丙烯酰胺10g与n,n-亚甲基双丙烯酰胺1g加入到混合离子溶液中,室温下磁力搅拌1h;随后将溶液置于50oc水浴锅中水浴搅拌0.5h,加入新制质量分数为30%的过硫酸铵溶液0.20ml,继续水浴搅拌6h形成湿凝胶;将湿凝胶置于80oc烘箱中干燥16h获干凝胶;将干凝胶转移至坩埚中于700oc的马弗炉中煅烧24h,随炉冷却至室温、研磨,即获得la1.38zr2o7:er0.6,yb0.02上转换发光材料。
本发明未详述之处,均为本技术领域技术人员的公知技术。最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!