甲基丙烯醛氧化制备甲基丙烯酸的催化剂及其制备方法与流程
本发明涉及一种用于甲基丙烯醛氧化制备甲基丙烯酸的催化剂,该催化剂属于复合氧化物催化剂类的非杂多酸催化剂,它不含多酸结构、具有良好的活性、选择性和稳定性,尤其是具有长的使用寿命。本发明还涉及该催化剂的制备方法及其在甲基丙烯醛氧化制备甲基丙烯酸反应中的应用。
背景技术:
甲基丙烯酸(maa)是一种重要的有机化工原料,广泛应用于生产有机玻璃、环保涂料、润滑剂及粘结剂等常用产品。由异丁烯氧化法制备maa的工艺,因原料来源广泛、原子利用率高、环境污染小等优点,目前备受关注。该工艺一般采用两步法合成工艺,将异丁烯在催化剂作用下部分氧化生成甲基丙烯醛(mal),然后mal在催化剂作用下进一步氧化生成maa。
在本领域,复合氧化物系指多组分氧化物,其中至少有一种是过渡金属氧化物。用于催化反应的复合氧化物即为复合氧化物催化剂。复合氧化物中,有的组分为主催化剂,有的组分明显作为载体,多数组分间要发生相互作用,形成相当复杂的结构,如杂多酸、含氧酸盐、尖晶石等复合氧化物,各种离子相互交叉的固济体以及单组分氧化物的各种混合物。其催化作用称为“配位络合电子迁移催化作用”。
杂多酸(polyoxometalates,简写为poms)是由杂原子(如p、si、fe、co等)和多原子(如mo、w、v、nb、ta等)按一定的结构通过氧原子配位桥联组成的一类含氧多酸,具有很高的催化活性,它不但具有酸性,而且具有氧化还原性,是一种多功能的新型催化剂。
mal氧化生成maa反应主要采用含磷钼的杂多酸化合物作为催化剂,对相关的杂多酸催化剂已有大量的研究,专利cn105517709a,us5856259,us5681790等表明,杂多酸化合物在对mal的氧化上具有良好的转化率以及对maa优异的选择性。
袁华等的“杂多酸催化剂”(《沧州师范专科学校学报》第23卷第2期,2007年6月)根据组成和结构将杂多酸催化剂分为五种类型,即keggin型(例如h3pw12o40)、anderson型(例如[temoo21]6-)、silver型(例如[cemo12o42]9-)、waugh型(例如(nh4)6[mnmo9o32]·8h2o)、和dawson型(例如k6[p2w18o62]·14h2o)。其中keggin型和dawson型结构是杂多酸化合物中最稳定的结构,相应的它们易合成,性质也稳定,因此对keggin型和dawson型杂多酸及其盐的研究较多也较充分。
目前常用的杂多酸化合物虽具有优良的活性及选择性,但高温易分解的特性是其致命的缺点,降低了催化剂的使用寿命及催化稳定性。因此,在mal通过氧化反应合成maa的反应中仍需要寻找一种在新的具有良好稳定性的催化剂。
中国专利cn1134296c公开了一种改进的催化剂载体和负载在载体上的钼-钒复合氧化物催化剂,该催化剂的组成为例如mo12v5w1cu2.2sb0.5。它并未涉及该催化剂的稳定性和使用寿命。
中国专利cn1153615c公开了一种复合氧化物催化剂,它具有通式mowbifeabcdeo,其中a为co或co+ni,b为碱金属元素、c为碱土金属元素、d选自p、te、sb、sn、ce、pb、nb、mn、as、b或zn,e选自si、al、ti或zr。该专利的发明目的是提供这样一种催化剂,它在活性、选择性和催化剂寿命方面是优良的,并且长期具有稳定性。在实施例部分其定性地给出如此结论,即“当催化剂用于反应多个小时后,观察到催化剂性能的微小降低”。
因此,仍需要寻找一种用于甲基丙烯醛氧化制备甲基丙烯酸的催化剂,它除了具有良好的mal转化率、maa选择性和maa收率以外,还至少能稳定运行一万小时或更长。
还需要寻找一种用于甲基丙烯醛氧化制备甲基丙烯酸的催化剂的制备方法。
技术实现要素:
本发明的目的是提供一种适用于甲基丙烯醛氧化合成甲基丙烯酸反应的非杂多酸体系催化剂,这种催化剂具有良好的活性、选择性以及稳定性。
本发明的另一个目的是提供一种所述催化剂的制备方法,通过本发明方法获得的催化剂能够以高转化率和高选择性来制备甲基丙烯酸。
因此,本发明的一个方面提供一种不含多酸结构的复合氧化物催化剂,它具有下式表示的组成:
mo12vatebnbcddoe
其中,
d表示至少一种碱金属元素;
a、b、c、d、e分别表示以mo12计,其它元素的摩尔数;
a=0.01-6.0;
b=0.01-4.0;
c=0.01-2.0;
d=0.2-3.0;
e为满足上述各组分的化合价所需要的氧的原子比率。
本发明的另一方面涉及本发明催化剂的制备方法,它包括如下步骤:
(i)根据所述催化剂的组成,将钼、钒、碲和d的前体化合物溶于溶剂中,配置a液;将铌的前体化合物溶于溶剂中,配置b液;
(ii)在35-95℃下,将a液和b液混合;
(iii)在含氧气体的流通下,于150-250℃预焙烧60~600分钟,得到催化剂前体;
(iv)对预焙烧后的催化剂成型,并在含氧气氛中在300-420℃焙烧120-1440分钟,含氧气体体积空速为200-1500h-1。
本发明的再一方面涉及本发明催化剂在甲基丙烯醛通过氧化反应合成甲基丙烯酸反应中的用途。
附图说明
下面结合附图进一步说明本发明,其中
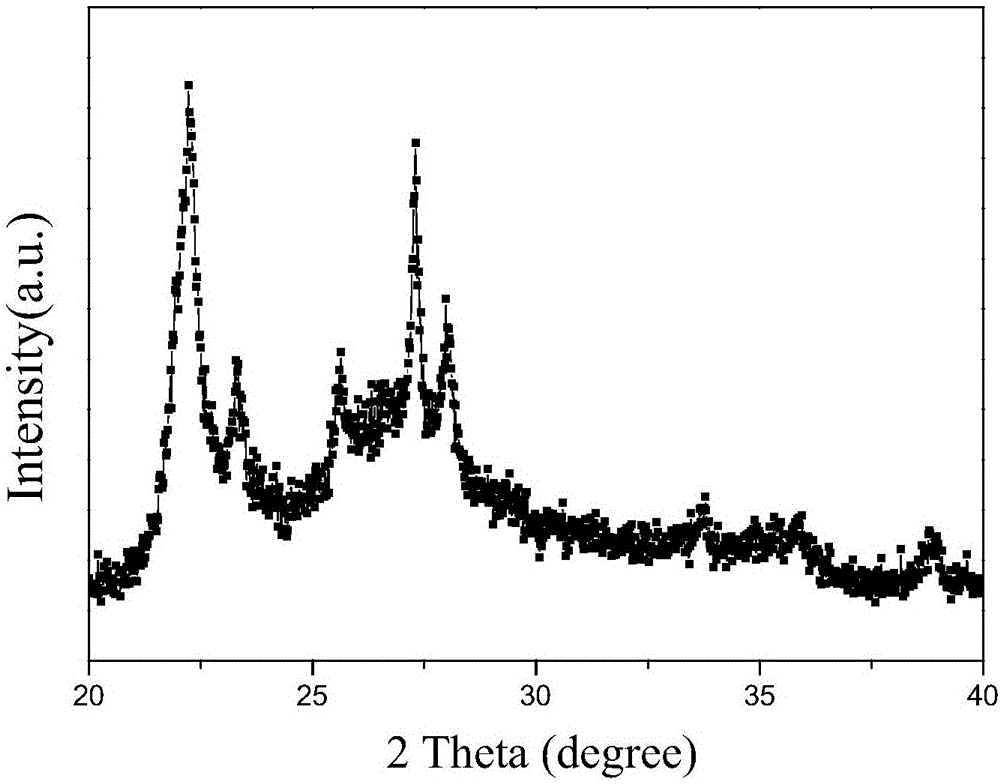
图1是实施例1催化剂的xrd图谱。
具体实施方式
1.催化剂
本发明提供一种催化剂,它具有下式表示的组成:
mo12vatebnbcddoe
mo、v、te、nb以及o分别表示钼、钒、碲、铌以及氧原子。
d表示选自锂(li)、钠(na)、钾(k)、铷(rb)或铯(cs)中至少一种碱金属元素;优选钠(na)、钾(k)或铯(cs)中至少一种元素。
a、b、c、d以及e表示各元素的原子比率:
a=0.01~6.0,更好为0.1~5.0,最好为0.3~4.0,优选0.5~3.0。
在本发明的一个较好实例中,a是由0.01、6.0、0.1、5.0、0.3、4.0、0.5和3.0中的任意两个作为端点形成的数值范围。
b=0.01~4.0,更好为0.1~3.0,最好为0.2~2.5,优选0.3~2.0。
在本发明的一个实例中,b是由0.01、4.0、0.1、3.0、0.2、2.5、0.3和2.0中的任意两个作为端点形成的数值范围。
c=0.01~2.0,更好为0.05~1.5,最好为0.1~1.25,优选0.3~1.0。
在本发明的一个实例中,b是由0.01、2.0、0.05、1.5、0.1、1.25、0.3和1.0中的任意两个作为端点形成的数值范围。
d=0.2~3.0,更好为0.3~2.5,最好为0.5~2.0,优选0.8~1.5。
在本发明的一个实例中,d是由0.2、3.0、0.3、2.5、0.5、2.0、0.8和1.5中的任意两个作为端点形成的数值范围。
e为满足上述各组分的化合价所需要的氧的原子比率。
在本发明的一个较好实例中,所述催化剂是复合氧化物催化剂。
在本发明中,术语“复合氧化物”是指不具有多酸结构的一类复合氧化物。
在本发明中,术语“不具有多酸结构”是指复合氧化物中多酸结构的含量小于10重量%,较好小于6重量%,更好小于4重量%,宜小于2重量%,优选小于1重量%。
在本领域中,术语“多酸”是指以mox(x值一般为6)为单元通过共角、共边(偶尔共面)氧联结缩聚成多金属氧酸化合物,即多酸化合物,更广义地称为金属-氧簇化合物(metal-oxygenclusters)。对于同多阴离子而言,m可以是其中一种或几种混合,m一般称(配原子)。对杂多阴离子也是一样,只是m可以被其他金属部分取代,另外杂多阴离子中还存在杂原子,周期表中大部分元素均可作为杂原子,因而杂多阴离子的种类很多,数目巨大。一般而言,多阴离子的结构是由通常处于最高氧化态(即d0,偶尔也有d1)的前过渡元素,以近八面体为基础。而后,通过共用氧原子,形成一类共角,共边和共面(极少)的结构独特的多核配合物。在了解了mo6八面体及八面体之间的连接方式之后,由这些结构单元就组成了多氧阴离子。例如,同多酸具有组成[mmoy]z-、杂多酸具有组成[xxmmoy]z-,m=w,mo,v,nb,ta等配原子、x为周期表中作为杂原子的70多种元素。
在本发明的一个实例中,所述催化剂选自mo12v3te1.5nb1k1o、mo12v3te1.5nb2k2o、mo12v3te1.5nb1cs1o、mo12v3te1.5nb2cs1或其两种或多种形成的混合物。
2.本发明催化剂的制备方法
本发明上述催化剂是采用如下方法制得的:将相应组分元素的前体化合物溶解,分别作为a液和b液;a、b两种料液配置好后,在35-95℃条件下将两种料液混合,从而制备含有上述全部催化剂成分的催化剂前体的悬浮分散浆液,并进行干燥,干燥后的催化剂前体经150-250℃预焙烧、300-420℃焙烧工序后得到成品催化剂。
(i)根据所述催化剂的组成,将钼、钒、碲和d的前体化合物溶于溶剂中,配置a液;将铌的前体化合物溶于溶剂中,配置b液;
所述a液通过将钼、钒、碲和d的前体化合物溶于溶剂中而配置。所述钼的前体化合物可以是,例如三氧化钼、仲钼酸铵或钼酸盐等,优选为仲钼酸铵。
所述钒的前体化合物可以是,例如五氧化二钒、偏钒酸铵等,优选偏钒酸铵。
所述碲的前体化合物可以是,例如二氧化碲、四氯化碲、碲酸等,优选碲酸。
所述d的前体化合物可以是,例如碱金属氧化物、氢氧化物、氯化物、硝酸盐、醋酸盐、含氧酸或含氧酸盐等,优选碱金属硝酸盐。
溶解前体化合物所需溶剂和温度没有特殊要求,只要使用的化合物能完全溶解或均匀分散即可,溶剂可列举例如水、甲醇、乙醇、乙醚、丙酮或其两种或更多种形成的混合物等,优选使用水。以制备浆料的前体化合物总量为100重量份计,水量约为100-1000重量份,优选100-500重量份。
b液通过将铌的前体化合物溶于溶剂中而配置,铌前体化合物包括,例如五氧化二铌、草酸铌、氢氧化铌等,优选草酸铌。溶解该前体化合物所需溶剂和温度没有特殊要求,只要使用的前体化合物能完全溶解或均匀分散即可,溶剂可列举例如水、甲醇、乙醇、乙醚、丙酮或其两种或更多种形成的混合物等,优选使用水。以制备浆料的前体化合物总量为100重量份计,水量约为100-500重量份,优选100-300重量份。
本发明催化剂中各活性组分的摩尔量,以12份钼原子为基准,钒的量为0.01-6.0份,较好为0.1-5.0份,更好为0.3-4.0份,优选0.5-3.0份。
其它活性组分的种类和使用量可根据催化剂的使用条件、所需性能等予以确定。
一般来讲,以12份钼原子为基准,元素te的使用量为0.01-4.0份,较好为0.1-3.0份,最好为0.2-2.5份,优选0.3-2.0份;
元素nb的使用量为0.01-2.0份,较好为0.05-1.5份,最好为0.1-1.25份,优选0.3-1.0份;
碱金属元素d的使用量为0.2-3.0份,更好为0.3-2.5份,最好为0.5-2.0份,优选0.8-1.5份。
(ii)在35~100℃下,将a液和b液混合;
制备混合浆料时,在35-95℃下将a液和b液混合,以得到高活性的催化剂。a液和b液的混合温度较好为40-90℃条件下混合,更好在45-80℃条件下混合,最好在50-70℃条件下混合。在本发明的一个较好实例中,所述混合温度是由35℃、95℃、40℃、90℃、45℃、80℃、50℃、70℃中的任意两个作为端点形成的温度范围。
对混料次序没有特殊要求,可以将a液加入b液中混合;也可将b液加入a液中混合。其中优选的方法是将b液加入a液中混合的方法。混合通常在搅拌的同时进行,得到均匀的悬浮浆液。
接下来,将上面获得的浆料干燥,对干燥方法和温度没有特殊要求,可以选择喷雾干燥,蒸发干燥,转鼓干燥等,优选喷雾干燥。
(iii)在含氧气体的流通下,于150-250℃预焙烧60-600分钟,得到催化剂前体。
本发明方法包括将得到的催化剂前体干燥物根据需要进行粉碎后,在空气等含氧气气体的流通下,于150-250℃预焙烧,优选180-220℃。预焙烧时间为60-600分钟,优选120-300分钟。
(iv)对预焙烧后的催化剂成型,并在含氧气氛中在300-420℃焙烧120-1440分钟,含氧气体体积空速为200-1500h-1。
本发明对催化剂成型方法没有特殊要求,可以使用已知的干式和湿式成型法,例如压片成型法、挤出成型法、造粒成型法等。对成型品的形状也没有特殊要求,可以选择圆柱形、环形、球形等所需要的形状。
催化剂焙烧需在含氧气的气氛下进行,焙烧温度选择为300-420℃,最好为320-410℃,优选380-400℃。焙烧时间为120-1440分钟,最好为240-1200分钟,优选300-720分钟。含氧气的气氛,氧气质量浓度不低于10%,最好不低于20%,优选空气。焙烧时,气体的流通速度以体积空速计,应为200-1500h-1,最好为400-1200h-1,优选500-1000h-1。
在本发明的一个实例中,所述催化剂的制造方法包括将钼酸铵、偏钒酸铵、碲酸、碱金属硝酸盐溶解于水中,得到溶液a;将草酸铌溶解于水中,得到溶液b。将a液升温至40-90℃,在搅拌条件下将b液倒入a液中,并继续搅拌得到含有催化剂前体的混合浆液。将此浆液在110-130℃下干燥,得到固体粉末。在空气气氛中200-250℃预焙烧,取固体粉末加入适量蒸馏水,挤出成型,制成10-20目的颗粒,之后在360-400℃空气流中焙烧制得最终的成品催化剂。
3.本发明催化剂的用途
通过上述方法制备的催化剂可用于气相氧化mal合成maa。在本发明的一个实例中,所述气相氧化反应包括如下步骤:原料mal、空气或含分子氧的稀释气体混合物及水蒸气经预热后,通入装有催化剂的固定床列管反应器中进行选择氧化反应合成maa。所用含分子氧的稀释气体混合物中,分子氧可以来自于纯氧、富氧或空气,稀释气体可以是n2、co、co2或h2o中的一种或它们按任意比例的混合物。
氧化反应条件为:温度240~300℃,优选250~290℃;压力0.05~0.5mpa,优选常压;反应原料混合气总空速600~5000h-1,优选1000~3000h-1;mal的摩尔浓度1~20%,优选3~10%;o2与mal的摩尔比1~8,优选2~4;水蒸气与mal的摩尔比2~10,优选3~8。
下面将用具体的实施例来说明高性能催化剂的制备方法,及其催化mal选择氧化生成maa的反应性能,但本发明的范围并不限于这些实施例。
实施例
在下列实施例中,使用如下公式计算mal氧化合成maa的转化率和选择性:
甲基丙烯醛转化率(%)=[(反应前甲基丙烯醛的量-反应后甲基丙烯醛的量)/反应前甲基丙烯醛的量]×100%
甲基丙烯酸的选择性(%)=(反应生成的甲基丙烯酸的量/反应的甲基丙烯醛的量)×100%
实施例1
1.催化剂的制备
将74.9克钼酸铵、12.3克偏钒酸铵、12.1克碲酸、3.5克硝酸钾溶解于500克蒸馏水中,得到溶液a;将18.8克草酸铌溶解于100克的蒸馏水中,得到溶液b。
将a液升温至40℃,在搅拌条件下将b液倒入a液中,并继续搅拌1小时,得到含有催化剂前体的混合浆液。
将此浆液在120℃下干燥24小时,得到固体粉末。在空气气氛中250℃预焙烧3小时,取80克固体粉末加入适量蒸馏水,挤出成型,制成10~20目的颗粒,之后在400℃空气流中焙烧3小时,空气空速1000h-1,制得最终的成品催化剂。此催化剂组成为mo12v3te1.5nb1k1o。其xrd图谱见附图1。
2.催化剂的性能评价
将25克10~20目的催化剂颗粒装入固定床管壳式反应器中,在280℃、常压、原料摩尔组成mal:o2:n2:h2o=1:2.4:8:10、空速1250h-1条件下进行选择氧化反应。反应进行48小时后采集反应产物进行气相色谱分析,反应结果见表1。
实施例2
1.催化剂的制备
将74.9克钼酸铵、12.3克偏钒酸铵、12.1克碲酸、7克硝酸钾溶解于500克蒸馏水中,得到溶液a;将37.6克草酸铌溶解于100克的蒸馏水中,得到溶液b。
将a液升温至40℃,在搅拌条件下将b液倒入a液中,并继续搅拌1小时,得到含有催化剂前体的混合浆液。
将此浆液在120℃下干燥24小时,得到固体粉末。在空气气氛中250℃预焙烧3小时,取80克固体粉末加入适量蒸馏水,挤出成型,制成10~20目的颗粒,之后在400℃空气流中焙烧3小时,空气空速1000h-1,制得最终的成品催化剂。此催化剂组成为mo12v3te1.5nb2k2o。
2.催化剂的性能评价
催化剂评价条件同实施例1,反应结果见表1。
实施例3
1.催化剂的制备
将74.9克钼酸铵、12.3克偏钒酸铵、12.1克碲酸、6.8克硝酸铯溶解于500克蒸馏水中,得到溶液a;将18.8克草酸铌溶解于100克的蒸馏水中,得到溶液b。
将a液升温至40℃,在搅拌条件下将b液倒入a液中,并继续搅拌1小时,得到含有催化剂前体的混合浆液。
将此浆液在120℃下干燥24小时,得到固体粉末。在空气气氛中250℃预焙烧3小时,取80克固体粉末加入适量蒸馏水,挤出成型,制成10~20目的颗粒,之后在400℃空气流中焙烧3小时,空气空速1000h-1,制得最终的成品催化剂。此催化剂组成为mo12v3te1.5nb1cs1o。
2.催化剂的性能评价
将20克10~20目的催化剂颗粒装入固定床管壳式反应器中,在280℃、常压、原料摩尔组成mal:o2:n2:h2o=1:2:8:10、空速1250h-1条件下进行选择氧化反应。反应进行48小时后采集反应产物进行气相色谱分析,反应结果见表1。反应10000小时后采集反应产物进行色谱分析,反应结果见表1。
实施例4
1.催化剂的制备
将74.9克钼酸铵、12.3克偏钒酸铵、12.1克碲酸、6.8克硝酸铯溶解于500克蒸馏水中,得到溶液a;将37.6克草酸铌溶解于100克的蒸馏水中,得到溶液b。
将a液升温至40℃,在搅拌条件下将b液倒入a液中,并继续搅拌1小时,得到含有催化剂前体的混合浆液。
将此浆液在120℃下干燥24小时,得到固体粉末。在空气气氛中250℃预焙烧3小时,取80克固体粉末加入适量蒸馏水,挤出成型,制成10~20目的颗粒,之后在400℃空气流中焙烧3小时,空气空速1000h-1,制得最终的成品催化剂。此催化剂组成为mo12v3te1.5nb2cs1。
2.催化剂的性能评价
催化剂评价条件同实施例2。反应进行48小时后采集反应产物进行气相色谱分析,反应结果见表1。反应进行10000小时后采集反应产物进行气相色谱分析,反应结果见表1。
表1反应结果
- 还没有人留言评论。精彩留言会获得点赞!