一种光催化制氢Z型光催化剂及其制备方法和应用与流程
本发明属于光催化领域,尤其涉及新型光催化剂的合成方法及催化剂在光催化制氢中的应用。
背景技术:
随着人口的增加和世界经济的快速发展,能源消耗将继续增加,能源短缺的时期将更早出现。煤炭,石油和天然气等传统化石燃料的大规模开发和利用导致这些不可再生能源载体迅速减少和消耗。探索绿色,高效和可持续替代的新能源载体已成为世界上大多数研究人员的热门话题。由于高燃烧值,无污染和低碳,氢气为能源领域带来了新的活力。一方面,太阳能资源丰富,免费使用,无运输成本,对环境无污染。另一方面,72%的地球表面被水覆盖。因此,通过使用太阳能来实现分解水产氢是一个必要和重要的研究课题。
自从fujishima和honda于1972年发现光电催化制氢从水分解以来,研究人员为探索高效稳定的光催化剂的研究做出了重要贡献。例如,通过掺杂金属离子和非金属离子来调节光催化剂的吸收范围。沉积在光催化剂表面上的助催化剂可以有效地分离光生电子和空穴,从而增强光催化活性。由n型和p型半导体组成的合适的p-n异质结由于能带位置关系产生的电位差可以有效地分离电荷。然而,水分解产生的光催化氢的效率仍然很低。其关键原因是光催化反应缺乏强大的驱动力,光源利用不足以及光生电子和空穴容易复合。2006年,报道了第一个全固态z型光催化剂。从那时起,z型光催化剂引起了广泛的关注。通过选择合适的半导体构建z型光催化剂,可以有效抑制光生电子和空穴的复合,拓宽光催化剂的光响应范围,提高光催化氧化还原反应的驱动能力。尽管z型光催化剂具有这些优点,但难以获得具有高活性的真正z型光催化体系。也就是说,在传统的制备方法中,例如,机械混合(m-m)方法,获得真正的z型光催化剂复合颗粒的数量非常少。而且,同种光催化剂颗粒的聚集占了很大比例,导致整个光催化体系的活性低。
技术实现要素:
为了解决电子和空穴的复合问题,本发明采用镀膜脱模(c-d)法合成了一种z型光催化剂(er3+:y3al5o12@nb2o5/pt/in2o3)用于有效分离光生电子和空穴的复合,pt作为导电通道能够加速电子转移,er3+:y3al5o12可以吸收可见光并将其转化为紫外光激活宽带隙nb2o5。最终合成的复合光催化剂er3+:y3al5o12@nb2o5/pt/in2o3,将其应用于光催化制氢中,具有很高的光催化制氢活性。
本发明采用的技术方案是:一种光催化制氢z型光催化剂,所述的光催化制氢z型光催化剂是er3+:y3al5o12@nb2o5/pt/in2o3。
优选的,上述的一种光催化制氢z型光催化剂,所述的er3+:y3al5o12@nb2o5/pt/in2o3中,pt的重量百分含量为0.5%;er3+:y3al5o12的重量百分含量为15.0%;按摩尔比,nb2o5:in2o3=1:1。
一种光催化制氢z型光催化剂的制备方法,包括如下步骤:
1)将er3+:y3al5o12@nb2o5溶胶,在1800转下通过旋涂法涂覆在模板上生长20-30s,80℃下干燥30-40min,重复三次,形成er3+:y3al5o12@nb2o5膜;
2)将氯酸铂溶液滴涂在er3+:y3al5o12@nb2o5膜上,通过离子吸附法将pt颗粒沉积在er3+:y3al5o12@nb2o5膜上,形成er3+:y3al5o12@nb2o5/pt膜;
3)将in2o3溶胶,在1800转下通过旋涂法涂覆在er3+:y3al5o12@nb2o5/pt膜上,生长20-30s,80℃下干燥30-40min,重复三次,形成er3+:y3al5o12@nb2o5/pt/in2o3膜;
4)将负载有er3+:y3al5o12@nb2o5/pt/in2o3膜的模板在400-600℃下,热处理1.0-3.0h,冷却后,脱模,研磨,获得目标产物er3+:y3al5o12@nb2o5/pt/in2o3。
优选的,上述的一种光催化制氢z型光催化剂的制备方法,所述的er3+:y3al5o12@nb2o5溶胶的制备方法包括如下步骤:将柠檬酸水溶液逐滴加入到nbcl5的乙醇溶液中,搅拌5-10min后加入er3+:y3al5o12继续搅拌30-40min,室温下陈化,形成er3+:y3al5o12@nb2o5溶胶。
优选的,上述的一种光催化制氢z型光催化剂的制备方法,所述的in2o3溶胶的制备方法包括如下步骤:在搅拌下,将去离子水、乙二醇和无水乙醇的混合溶液逐滴加入到incl3的冰醋酸溶液中,搅拌均匀后,加入柠檬酸,将所得混合溶液在50℃下回流2-3h,冷却至室温并老化,得in2o3溶胶。
上述的z型光催化剂在光催化制氢中的应用。方法如下:于含有甲醇的水溶液中,加入上述的光催化制氢z型光催化剂er3+:y3al5o12@nb2o5/pt/in2o3,在温度25℃和压力101325pa下,用300w的氙灯照射300min。
本发明的光催化制氢z型光催化剂er3+:y3al5o12@nb2o5/pt/in2o3在可见光照射下光催化分解水制氢的过程分析:z型光催化体系由in2o3,er3+:y3al5o12@nb2o5和pt纳米粒子组成,其中pt纳米粒子位于in2o3和er3+:y3al5o12@nb2o5之间。由于in2o3的价带相对接近电势为nb2o5的导带,理想的z型光催化体系可由in2o3和nb2o5组成。特别是nb2o5导带上的电子可以通过pt纳米粒子进入in2o3的价带,然后与空穴复合,有效地抑制nb2o5和in2o3中光生电子和空穴的复合。当z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂被可见光照射时,由于其带隙窄,in2o3可以被可见光直接激发以形成光生电子和空穴。此外,可见光通过er3+:y3al5o12转换为高能紫外光,激发宽带隙nb2o5,产生光生电子和空穴。由于in2o3的价带相对接近电势为nb2o5的导带,因此nb2o5导带上的电子可以通过pt纳米粒子进入in2o3的价带,并与空穴复合。in2o3导带上的电子和nb2o5价带上的空穴分别进行氧化还原反应。nb2o5价带上的空穴可以氧化甲醇作为牺牲剂,促进in2o3导带上的电子还原为氢。因此,可以看出,在甲醇存在下,z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂可以有效地进行光催化制氢。
本发明的有益效果:
本发明制备的复合光催化剂er3+:y3al5o12@nb2o5/pt/in2o3不仅具有传统z型光催化剂的优点,而且更值得关注的是本发明提出了一种新的制备方法,即涂层脱模(c-d)方法,用于获得高比例的z型光催化剂颗粒。基于z型光催化剂的优点,通过这种强制性方法有效结合不同催化剂颗粒可以大大提高高活性z型光催化剂复合颗粒的比例,并减少非z型复合颗粒的数量。反过来,可以提高整个光催化系统的活性。另外,通过该方法可以将一些作为导电通道的贵金属颗粒强制固定在两个光催化剂颗粒之间,以形成改进的z型光催化体系。
附图说明
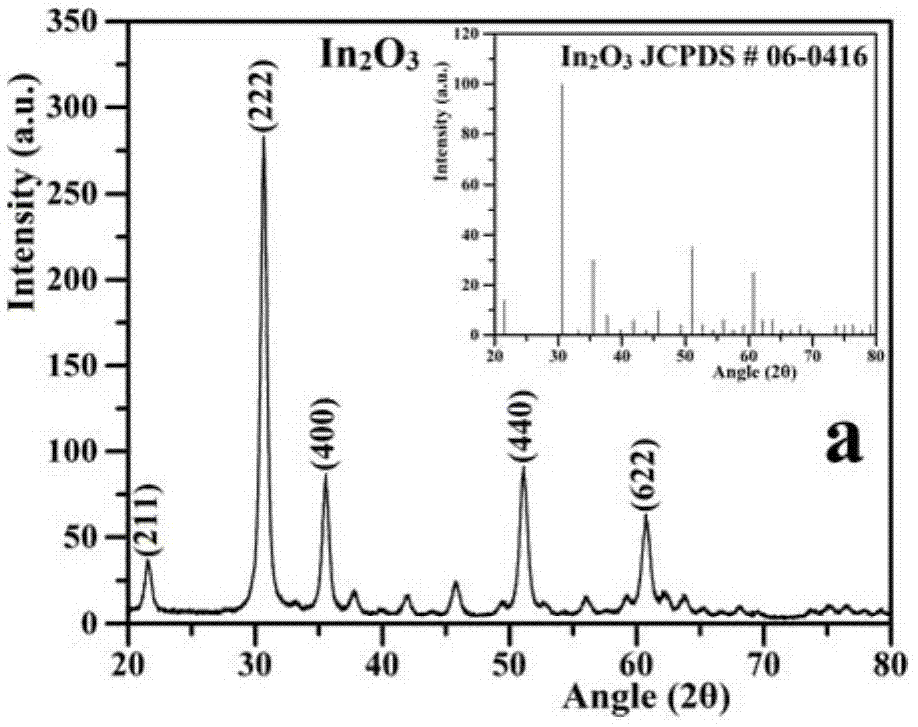
图1a是in2o3的x射线粉末衍射(xrd)图。
图1b是nb2o5的x射线粉末衍射(xrd)图。
图1c是er3+:y3al5o12的x射线粉末衍射(xrd)图。
图1d是er3+:y3al5o12@nb2o5/in2o3(m-m)的x射线粉末衍射(xrd)图。
图1e是er3+:y3al5o12@nb2o5/in2o3(c-d)的x射线粉末衍射(xrd)图。
图1f是er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的x射线粉末衍射(xrd)图。
图2是in2o3,nb2o5,er3+:y3al5o12和er3+:y3al5o12@nb2o5/pt/in2o3的扫描电子显微镜(sem)图。
图3是er3+:y3al5o12@nb2o5/pt/in2o3的透射电子显微镜(tem)图。
图4是er3+:y3al5o12@nb2o5/pt/in2o3的x射线能量色散光谱(edx)图。
图5a是er3+:y3al5o12@nb2o5/pt/in2o3的x射线光电子能谱(xps)图。
图5b是in(3d)的x射线光电子能谱(xps)图,
图5c是nb(3d)的x射线光电子能谱(xps)图。
图5d是er(4d)的x射线光电子能谱(xps)图。
图5e是y(3d)的x射线光电子能谱(xps)图。
图5f是al(2p)的x射线光电子能谱(xps)图。
图5g是o(1s)的x射线光电子能谱(xps)图。
图5h是pt(4f)的x射线光电子能谱(xps)图。
图6a是in2o3,nb2o5和er3+:y3al5o12@nb2o5/in2o3的uv-vis漫反射光谱(drs)图。
图6b是in2o3的带隙估算图。
图6c是nb2o5的带隙估算图。
图7a是in2o3的傅立叶变换红外光谱(ft-ir)图。
图7b是nb2o5的傅立叶变换红外光谱(ft-ir)图。
图7c是er3+:y3al5o12的傅立叶变换红外光谱(ft-ir)图。
图7d是er3+:y3al5o12@nb2o5/pt/in2o3的傅立叶变换红外光谱(ft-ir)图。
图8是in2o3,nb2o5,er3+:y3al5o12@nb2o5/in2o3(m-m),er3+:y3al5o12@nb2o5/in2o3(c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的光致发光(pl)光谱图。
图9a是er3+:y3al5o12@nb2o5/in2o3(m-m),er3+:y3al5o12@nb2o5/in2o3(c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的电化学测试。
图9b是er3+:y3al5o12@nb2o5/in2o3(m-m),er3+:y3al5o12@nb2o5/in2o3(c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的电化学测试。
图10a是不同方法制备催化剂对光催化制氢的影响图。
图10b是不同热处理温度制备催化剂对光催化制氢的影响图。
图10c是不同热处理时间制备催化剂对光催化制氢的影响图。
图10d是使用次数对光催化剂er3+:y3al5o12@nb2o5/pt/in2o3光催化制氢活性的影响图。
图11是z型光催化剂er3+:y3al5o12@nb2o5/pt/in2o3光催化制氢的机理图。
具体实施方式
实施例1一种光催化制氢z型光催化剂er3+:y3al5o12@nb2o5/pt/in2o3
(一)制备方法
1、制备er3+:y3al5o12粉末
在搅拌下,将2.27gy2o3和0.01ger2o3溶解在适当的热hno3溶液中,得到y(no3)3和er(no3)3的混合溶液。然后,在磁力搅拌下加入12.62gal(no3)3·9h2o,并获得均匀溶液。之后,将33.93g固体柠檬酸也加入到所述混合物溶液中。继续搅拌溶液并在50-60℃下加热直至成功形成透明溶胶。随后,将溶胶在80℃下加热24h,并成为干凝胶。将凝胶在空气中冷却后,将其研磨成均匀的粉末。将得到的前体在马弗炉中在1100℃下煅烧2.0h,以除去残留的有机组分和硝酸根离子。最后,使烧结物质在空气中冷却至室温。完全冷却后,获得er3+:y3al5o12纳米尺寸的颗粒。
2、制备er3+:y3al5o12@nb2o5溶胶
将7.4gnbcl5溶于10.0ml无水乙醇中,并在70℃下搅拌30min,将其标记为溶液a。将17.2g柠檬酸溶于10.0ml去离子水中,将其标记为溶液b。将溶液b逐滴添加到溶液a中,搅拌5-10min后加入0.5ger3+:y3al5o12继续搅拌30-40min,室温下陈化,形成er3+:y3al5o12@nb2o5溶胶。
3、制备in2o3溶胶
将8.0gincl3加入10.0ml冰醋酸中,然后剧烈搅拌30min,将其标记为溶液a。将3.0ml去离子水和2.0ml乙二醇加入5.0ml无水乙醇中,然后搅拌30min,其标记为溶液b。在搅拌下,将溶液b缓慢滴入溶液a中。最后,将1.2g柠檬酸加入混合物溶液中。将所得溶液转移至三颈烧瓶中,并在50℃下回流2.5h。将溶液在室温下冷却并老化,得到浅黄色in2o3溶胶。
4、机械混合法(m-m)制备er3+:y3al5o12@nb2o5/in2o3
将20mgin2o3溶胶直接与20mger3+:y3al5o12@nb2o5溶胶混合,超声分散1.0h。然后,将混合溶胶加热,在80℃下干燥24h,形成干凝胶。将干凝胶在空气中冷却后,磨成均匀的粉末。将粉末于500℃下热处理2.0h,冷却,研磨,获得z型er3+:y3al5o12@nb2o5/in2o3光催化剂。
5、镀膜-脱模法(c-d)制备er3+:y3al5o12@nb2o5/in2o3
将er3+:y3al5o12@nb2o5溶胶,在1800转下通过旋涂法涂覆在玻璃基板上生长20s,80℃下干燥30min,旋涂过程重复三次,得er3+:y3al5o12@nb2o5膜。之后,再将in2o3溶胶,在1800转下通过旋涂法在er3+:y3al5o12@nb2o5膜上生长20s,在80℃下干燥30分钟。旋涂过程重复三次。将负载有er3+:y3al5o12@nb2o5/in2o3膜的玻璃基板在500℃热处理2.0h。最后,通过脱模,研磨处理获得z型er3+:y3al5o12@nb2o5/in2o3光催化剂。
6、镀膜-脱模法(c-d)制备er3+:y3al5o12@nb2o5/pt/in2o3
将er3+:y3al5o12@nb2o5溶胶,在1800转下通过旋涂法涂覆在玻璃基板上生长20s,80℃下干燥30min。旋涂过程重复三次,形成er3+:y3al5o12@nb2o5膜。
将氯酸铂溶液滴涂在er3+:y3al5o12@nb2o5膜上,通过离子吸附法将pt颗粒沉积在er3+:y3al5o12@nb2o5膜上,形成er3+:y3al5o12@nb2o5/pt膜。
将in2o3溶胶,在1800转下通过旋涂法涂覆在er3+:y3al5o12@nb2o5/pt膜上,生长20s,80℃下干燥30分钟,重复三次,形成er3+:y3al5o12@nb2o5/pt/in2o3膜。
将负载有er3+:y3al5o12@nb2o5/pt/in2o3膜的玻璃基板分别在不同温度(400℃,500℃和600℃)和不同时间(1.0h,2.0h和3.0h)下进行热处理。冷却后,脱模,研磨,分别获得不同处理温度和不同处理时间的目标产物er3+:y3al5o12@nb2o5/pt/in2o3。
(二)检测
(1)in2o3,nb2o5,er3+:y3al5o12,er3+:y3al5o12@nb2o5/in2o3(m-m和c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的x射线粉末衍射(xrd)
通过x射线衍射(xrd)研究了in2o3,nb2o5,er3+:y3al5o12,er3+:y3al5o12@nb2o5/in2o3(m-m和c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的晶体结构,结果如图1a-图1f所示。图1a显示了在2θ=21.75°,30.66°,35.47°,51.12°和60.73°处的衍射峰,它们分别对应于in2o3晶格平面((211),(222),(400),(440)和(622))。良好结晶的衍射峰与立方结构类型in2o3的标准卡jcpds#06-0416数据一致。如图1b所示,合成的nb2o5的衍射峰分别位于22.68°(001),28.61°(180),36.75°(181),46.30°(002),50.59°(380)和55.31°(202),与nb2o5标准卡jcpds#30-0873数据一致。xrd图显示制备的样品是高纯度的nb2o5纳米颗粒,并且没有任何其他价态的氧化铌相(例如nbo和nbo2)。在图1c中,合成的er3+:y3al5o12的主要衍射峰位于27.77°(321),29.72°(400),33.34°(420),36.62°(422),41.14°(521),46.56°(532),55.09°(640)和61.78°(800),与y3al5o12的标准卡jcpds#33-0040数据几乎相同。尖峰表明已成功合成具有非常高结晶度的er3+:y3al5o12。然而,对于一些主要的衍射峰,与y3al5o12的标准卡数据相比有轻微的移动,这表明在制备过程中,少量的er3+离子已经取代了一部分y3+离子,从而进入了y3al5o12的内部晶格。图1d和图1e分别显示了m-m法和c-d法制备的er3+:y3al5o12@nb2o5/in2o3的xrd谱。从xrd图谱可以清楚地看到er3+:y3al5o12,in2o3和nb2o5的特征衍射峰,表明通过m-m和c-d制备方法制备的样品含有这些氧化物粒子。然而,尽管通过m-m方法制备的样品给出了in2o3(30.66°)和nb2o5(28.61°)的特征峰,但这些特征衍射峰相对独立,并且几乎与纯in2o3和nb2o5的峰相同。这表明大多数in2o3和nb2o5没有紧密结合在一起。对于c-d法制备的er3+:y3al5o12@nb2o5/in2o3,在图1e中,弱衍射峰出现在28.61°,属于解离的nb2o5。此外,新的强和宽的衍射峰出现在30.52°,这可能是由于in2o3和nb2o5的衍射峰的相对位移,表明in2o3和nb2o5紧密结合在一起。在图1f中,可以找到与图1e中相同的特性。此外,pt的特征峰也可分别在39.83°,46.48°和68.03°位置处看到。它们与pt标准卡jcpds#01-1194数据一致。而且,所有特征衍射峰都是具有高强度尖锐的峰。表面c-der3+:y3al5o12@nb2o5/pt/in2o3具有非常高的结晶度。并且,in2o3和nb2o5可以紧密结合在一起,pt纳米尺寸的颗粒位于in2o3和nb2o5之间,形成高比例的改性z型光催化剂。
(2)in2o3,nb2o5,er3+:y3al5o12和er3+:y3al5o12@nb2o5/pt/in2o3的扫描电子显微镜(sem)
in2o3(a),nb2o5(b),er3+:y3al5o12(c)和er3+:y3al5o12@nb2o5/pt/in2o3的不同放大倍数1:1.0μm(d-1),2:500(d-2)nm和3:200nm(d-3)的形貌和结构通过sem评估(图2)。在图2的(a)中,in2o3颗粒呈现立方结构,尺寸范围为200-300nm。在图2的(b)中,nb2o5样品显示出具有大尺寸和规则层状结构的聚集体形态。作为上转换发光剂的er3+:y3al5o12的形态如图2的(c)所示。显然,er3+:y3al5o12颗粒是一些长轴为50nm,短轴为30nm的椭圆体。图2的(d)显示了通过c-d方法制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的不同尺度的sem图像。从图2的(d-1(1.0μm))可以看出,er3+:y3al5o12@nb2o5/pt/in2o3纳米颗粒聚集在一起。这些聚集体的粒径为约1.0μm。在图2的(d-2(500nm))中,每个聚集的er3+:y3al5o12@nb2o5/pt/in2o3颗粒由相对小的in2o3和er3+:y3al5o12@nb2o5颗粒组成。进一步扩大比例,在图2(d-3(200nm))中,可以清楚地看出in2o3和er3+:y3al5o12@nb2o5紧密结合。此外,在in2o3表面上存在一些微小颗粒,可以确认为pt纳米颗粒。这也表明许多pt纳米尺寸的颗粒应位于er3+:y3al5o12@nb2o5和in2o3之间。从sem图像判断,本发明成功地采用c-d法制备了z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂。
(3)er3+:y3al5o12@nb2o5/pt/in2o3的透射电子显微镜(tem)
通过tem和高分辨率tem(hrtem)进一步进行深入观察,如图3所示。从图3中(a)可以看出,制备的光催化剂由四部分组成。其中,较暗的部分是nb2o5,并且在nb2o5颗粒的中间具有不规则球形的黑色部分是er3+:y3al5o12纳米颗粒。较浅的颜色部分是in2o3。另外,in2o3和nb2o5颗粒之间的小的深色球是pt纳米尺寸的颗粒。此外,如图3中(b)所示,通过hrtem观察er3+:y3al5o12@nb2o5/pt/in2o3中四个部分的晶体结构,并通过测量晶格间距确认它们的关系。在图像的中间,可以看到一组0.390nm的晶格条纹,其对应于nb2o5的(001)面(jcpds#30-0873)。图像右上角的0.295nm晶格条纹归于立方in2o3的(222)平面(jcpds#06-0416)。此外,在图像中也观察到er3+:y3al5o12的0.265nm晶格条纹,这归因于y3al5o12的(420)晶格面(jcpds#33-0040)。与图像中的nb2o5相邻的0.198nm晶格条纹属于pt的(200)面(jcpds#01-1194)。从图3中可以看出,可以进一步证实z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的组成和四个部分的相互关系。
(4)er3+:y3al5o12@nb2o5/pt/in2o3的x射线能量色散光谱(edx)图
为了确定制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的元素组成,研究了其能量色散x射线光谱(edx)。如图4所示,制备的er3+:y3al5o12@nb2o5/pt/in2o3由er,y,al,nb,pt,in和o元素组成。可以认为in,nb和o元素的强特征峰存在于in2o3和nb2o5中。er,y,al和o元素的特征峰证实er3+:y3al5o12作为上转换发光剂已成功加入到制备的z型光催化剂中。此外,还可以发现pt原子的衍射峰,这进一步证实了pt原子的存在。这些观察结果与xrd,sem和tem的结果基本一致。
(5)er3+:y3al5o12@nb2o5/pt/in2o3,in(3d),nb(3d),er(4d),y(3d),al(2p),o(1s)和pt(4f)的x射线光电子能谱(xps)图
通过x射线光电子能谱(xps)研究了z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的基本组成和价键结构,如图5a-图5h所示。其中,图5a显示了制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的xps光谱。这表明制备的样品含有er,y,al,nb,pt,in和o元素。在图5b中在444.6ev和452.1ev观察到的峰可分别归因于in3d5/2和in3d3/2,它们是三价铟离子的特征氧化态。图5c中高分辨率的nb3d谱中的两个特征峰分别为207.7ev和210.46ev,可分别归属于nb3d5/2和nb3d3/2自旋轨道峰,表明所有铌原子都以nb5+的形式存在于这种复合材料中。图5d,图5e,图5f和图5g表示168.3ev(er4d5/2),158.1ev(y3d5/2),160.1ev(y3d3/2),74.3ev(al2p3/2),74.5ev(al2p1/2)和531.4ev(o1s),它们与er3+:y3al5o12的组成密切相关。图5g显示了氧物质的三个峰。位于530.6ev的第一个峰归因于o-nb键,而位于531.4ev的第二个峰可能是in2o3的晶格氧。在532.2ev观察到的第三个峰可归属于吸附的氧原子。在图5h中,pt4f的两个峰可以分别在71.3ev和74.0ev的结合能下发现,证实了金属铂的存在。
(6)in2o3,nb2o5和er3+:y3al5o12@nb2o5/pt/in2o3紫外-可见漫反射光谱(drs)与in2o3和nb2o5的带隙估算图
通过uv-vis漫反射光谱测定in2o3,nb2o5和er3+:y3al5o12@nb2o5/pt/in2o3的光学吸收,并使用基于kubelka-munk函数的吸光度与波长数据计算in2o3和nb2o5的带隙。从图6a可以看出,in2o3在450nm处显示出吸收边缘,这证明制备的in2o3可以吸收部分可见光。nb2o5显示了355nm的吸收带边缘,这表明制备的nb2o5只能吸收紫外光。由于er3+:y3al5o12的存在,er3+:y3al5o12@nb2o5/pt/in2o3的吸收光谱尾部在450-800nm处明显升高。该结果表明,er3+:y3al5o12可以吸收可见光并将它们转换成紫外光,以激活宽带隙nb2o5半导体,如图6a所示。图6b和图6c,根据kubelka-munk((αhν)2=c(hν-eg))公式,通过外推(αhν)2的线性部分与能量图来估计in2o3和nb2o5的带隙。其中α,c,ν和h分别是吸光度,常数,频率和普朗克常数。计算得到的in2o3和nb2o5的带隙分别约为2.82ev和3.32ev,与先前报道的值相似。
(7)in2o3,nb2o5,er3+:y3al5o12和er3+:y3al5o12@nb2o5/pt/in2o3的傅立叶变换红外光谱(ft-ir)
为了证明样品中键合结构的化学信息,制备的in2o3,nb2o5,er3+:y3al5o12和er3+:y3al5o12@nb2o5/pt/in2o3的ft-ir光谱如图7a-图7d所示。从图7a中可以看出在540cm-1,565cm-1和600cm-1处出现三个尖锐的吸收峰可以归因于in-o键的声子振动,这表明形成了立方in2o3。在图7b中,nb-o基团的两个非常宽的拉伸振动吸收带分别在642cm-1和864cm-1,表明nb2o5样品已经制备好。图7c给出了制备的er3+:y3al5o12的ft-ir光谱。在482cm-1,512cm-1和567cm-1处的吸收峰属于al-o6-al的拉伸和弯曲振动,在698cm-1和793cm-1处的吸收峰是拉伸和弯曲的al-o4-al的振动和729cm-1处的吸收峰对应于y-o键。在图7d中,可以发现in2o3,nb2o5和er3+:y3al5o12的所有特征吸收峰,但是一些峰具有轻微的运动和增强。可以推断,in2o3和nb2o5结合在一起且er3+:y3al5o12被nb2o5包覆。同时,可以确认3400cm-1附近的宽吸收带对应于-oh氢键的伸缩振动。在1600cm-1处的明显吸收可归因于-oh基团吸附的水分子的变形振动。
(8)in2o3,nb2o5,er3+:y3al5o12@nb2o5/in2o3(m-m),er3+:y3al5o12@nb2o5/in2o3(c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的光致发光(pl)光谱
为了研究电荷分离和转移特性,制备的样品进行了pl测试,结果显示在图8中。众所周知,高的光催化活性是由于光生电子-空穴对的有效分离。简单地说,光催化剂的低pl发射强度表明光生电子和空穴的复合率低,这反过来表现出高的光催化活性。当制备的in2o3和nb2o5在310nm波长的光下被激发时,两个强pl发射峰出现在470nm处,这表明in2o3和nb2o5具有更高的光生电子和空穴的复合率。与in2o3和nb2o5相比,通过两种不同(m-m和c-d)方法制备的z型光催化剂在相同的光激发波长下均显示出相对低的pl发射强度。这表明z型光催化剂的光生电子和空穴的复合率显着降低。值得注意的是,采用c-d法制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂在所有样品中具有最低的pl发射强度。一方面,c-d制备方法可有效提高高活性z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂颗粒的比例。另一方面,nb2o5导带上的电子可以通过pt纳米颗粒快速转移到in2o3的价带,与空穴重新结合。这有效地抑制了光生成的in2o3和nb2o5的电子-空穴对的复合。因此z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂可以显示出高的光催化活性。
(9)er3+:y3al5o12@nb2o5/in2o3(m-m),er3+:y3al5o12@nb2o5/in2o3(c-d)和er3+:y3al5o12@nb2o5/pt/in2o3(c-d)的电化学测试
光电化学测量通常用于定性研究光催化中光生电荷载体的激发和转移。在图9a中,为了解制备的光催化剂中的电子传输行为,电化学阻抗谱结果以奈奎斯特图的形式呈现。较小的电弧尺寸通常意味着光催化剂的表面具有低的电荷转移电阻。显然,用c-d法制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的弧半径最小。换句话说,z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂具有最低的界面电荷转移电阻,导致电子和空穴的高分离效率。相比之下,通过m-m法制备的z型er3+:y3al5o12@nb2o5/in2o3光催化剂具有最大的弧半径,因此,它具有最高的界面电荷转移电阻,导致电子和空穴的低分离效率。
为了进一步了解这些通过不同方法制备的光催化体系中的光生电荷分离效率和电子转移行为,测试了制备的光催化剂的瞬态光电流响应。如图9b所示,所有在开灯和关灯时的样品在可见光照射下显示相对稳定和可逆的光响应,每20秒重复一次,偏压电位为+0.62v。当灯亮时,光电流急剧增加,然后在灯灭之后立即返回其原始状态。通过m-m法制备的z型er3+:y3al5o12@nb2o5/in2o3光催化剂表现出非常低的光电流。并且,由于不存在pt纳米颗粒作为电子通道,c-d法制备的z型er3+:y3al5o12@nb2o5/in2o3也表现出相对低的光电流。然而,通过c-d法制备的z型er3+:y3al5o12@nb2o5/pt/in2o3显示出非常强的光电流。该结果进一步证实,通过使用c-d方法可以大大提高高活性z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂颗粒的比例。它导致整体光催化活性的增强。相比之下,c-d法制备的z型er3+:y3al5o12@nb2o5/pt/in2o3的光电流远高于c-d法制备的er3+:y3al5o12@nb2o5/in2o3(没有pt作为导电通道)的光电流。这是因为pt纳米颗粒的存在使得nb2o5导带上的电子能够快速转移到in2o3的价带,然后与空穴重新结合。显然,光电流分析与pl和电化学阻抗谱的结果一致。
实施例2er3+:y3al5o12@nb2o5/pt/in2o3在光催化制氢中的应用
实验方法:光催化氢生成实验在500mlpyrex反应器系统中,在温度25℃和压力101325pa下进行。在用于氢发生器的典型光催化反应中,将制备的样品在恒定搅拌下在含有甲醇的水溶液中放入光反应器中。在可见光照射之前,将反应系统通氩气30分钟以除去溶解的空气。然后通过来自300wxe灯(lx-300,deruifeng硬件电器企业,中国)的截止滤光器用可见光照射该系统4.0小时,该灯环绕在光反应器的侧面。通过冷镜(cm-1)和水过滤器的组合来控制照射光波长。对于可见光照射,将滤光片(l42)装配到上述光源(420<λ<800nm)。
(一)不同方法制备催化剂对光催化制氢的影响
不同制备方法对制备的z型光催化剂的制氢活性的影响如图10a所示。可以发现,这三种z型光催化剂的氢产生量均随着照射时间的增加而增加。然而,两个用c-d法制备的z型光催化剂的氢产生量远高于用m-m法制备的z型光催化剂的产氢量。这表明c-d方法可以大大提高高活性z-型光催化剂复合颗粒的比例,并减少非z-型光催化剂颗粒作为副产物的数量。也就是说,c-d方法可以提升z型光催化系统的活性。对于c-d法制备的z型光催化剂,有必要指出的是,pt纳米颗粒作为导电通道的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的产氢量远远高于z型er3+:y3al5o12@nb2o5/in2o3。这是因为pt纳米粒子作为导电通道通过c-d方法被强制固定在nb2o5和in2o颗粒之间,形成有效的z型光催化体系。因此,在这种z型er3+:y3al5o12@nb2o5/pt/in2o3光催化体系中,nb2o5导带上的电子可以快速转移到in2o的价带中,与空穴结合。综上所述,采用c-d方法可以得到高活性的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化体系,pt纳米粒子的存在可以加速光生电子和空穴的分离。
(二)不同热处理温度制备催化剂对光催化制氢的影响
图10b显示了热处理温度对制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的制氢活性的影响。可以看出,对于三个热处理温度,z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的氢产量均随着照射时间的增加而增加。然而,随着热处理温度的增加,在任何照射时间,氢产量都明显地增加(从400℃到500℃)然后略微减少(从500℃到600℃)。当z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂在500℃下煅烧2.0h时,可见光催化制氢的量达到最大值。因此,适当的高温有利于形成z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂。但是,即使温度进一步升高,制备的光催化剂的活性也几乎不变。可以证实,500℃是制备z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的最佳热处理温度,产氢量可达384μmol·g-1。
(三)不同热处理时间制备催化剂对光催化制氢的影响
图10c显示了热处理时间对制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的光催化制氢活性的影响。类似地,三个热处理时间的z型光催化剂的氢产量均随着照射时间的增加而增加。此外,可以发现,当z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂在500℃下煅烧1.0h时,它显示出最高的光催化活性。也就是说,随着热处理时间的增加,在任何照射时间的氢产量都减少。这表明,对于z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂,仅在1.0h热处理后才能获得高活性。而且,经过较长时间的热处理后,光催化活性会略微下降。这是因为在相对高的温度下,长期热处理会引起pt纳米颗粒的聚集。因此,对于500℃温度,1.0h的煅烧是形成高活性z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的合适热处理时间,产氢量可达410μmol·g-1。这不仅有助于提高z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的光催化活性,而且还避免了较长的热处理时间。
(四)使用次数对光催化剂er3+:y3al5o12@nb2o5/pt/in2o3光催化制氢活性的影响
最后,使用时间对制备的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的光催化制氢活性的影响如图10d所示。对于光催化体系,高光催化活性是评估光催化剂的关键。此外,光催化剂的使用时间是评价性能稳定性的另一个重要因素。在这部分中,每个循环实验的时间为4.0h,然后在80℃下热处理1.0h以进行活化后,回收光催化剂以进行相同的循环实验。可以看出,在每次重复实验后,z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的氢产量略有下降,但仍保持高水平。当然,在通过c-d法制备高比例的z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂的同时,需要一些适当的改进以保持稳定的高光催化活性。
er3+:y3al5o12@nb2o5/pt/in2o3光催化制氢的原理为:如图11所示,z型光催化体系由in2o3,er3+:y3al5o12@nb2o5和pt纳米粒子组成,其中pt纳米粒子位于in2o3和er3+:y3al5o12@nb2o5之间。由于in2o3的价带相对接近电势为nb2o5的导带,理想的z型光催化体系可由in2o3和nb2o5组成。特别是nb2o5导带上的电子可以通过pt纳米粒子进入in2o3的价带,然后与空穴复合,有效地抑制nb2o5和in2o3中光生电子和空穴的复合。当z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂被可见光照射时,由于其带隙窄,in2o3可以被可见光直接激发以形成光生电子和空穴。此外,可见光通过er3+:y3al5o12转换为高能紫外光,激发宽带隙nb2o5,产生光生电子和空穴。由于in2o3的价带相对接近电势为nb2o5的导带,因此nb2o5导带上的电子可以通过pt纳米粒子进入in2o3的价带,并与空穴复合。in2o3导带上的电子和nb2o5价带上的空穴分别进行氧化还原反应。nb2o5价带上的空穴可以氧化甲醇作为牺牲剂,促进in2o3导带上的电子还原为氢生成。因此,可以看出,在甲醇存在下,z型er3+:y3al5o12@nb2o5/pt/in2o3光催化剂可以有效地进行光催化制氢。
- 还没有人留言评论。精彩留言会获得点赞!
- 可见光响应的光催化剂Li<sub>5</sub>Cu<sub>8</sub>In<sub>5</sub>Ge<sub>9</sub>O<sub>36</sub>及其制备方法
- 具有还原Cr(VI)离子性质的硒醚亚铜簇负载型可见光催化剂的制作方法
- 一种对双酚a具有高矿化率的可见光响应型复合光催化剂的制作方法
- 多级式光催化臭氧氧化反应器及其光催化剂的制备方法
- 一种z型光催化剂及其制备方法
- 抗病毒性组合物、抗病毒剂、光催化剂以及病毒灭活方法
- 一种复合可见光光催化剂Ag<sub>2</sub>CO<sub>3</sub>/TiO<sub>2</sub>/ UiO-66-(COOH)<sub>2</sub>的制备方法及其应用
- 一种石墨烯负载的具有凹面立方体形貌的Ag光催化剂的制备方法
- 一种选择性专一识别的PPyZnFe2O4磁性印迹复合光催化剂的制备方法
- 的制备方法及应用