一种钌和钨改性的金属固溶体催化剂、其制备方法和解聚木质素的方法与流程
本发明涉及木质素纤维素类生物质转化技术领域,尤其是涉及一种钌和钨改性的金属固溶体催化剂、其制备方法和解聚木质素的方法。
背景技术:
生物质是公认的可再生有机碳的最佳来源;生物质主要由纤维素、半纤维素、木质素这三大组分组成。其中,木质素约占生物质三大组分的三分之一,但是与纤维素与半纤维素不同,木质素并没有被很好地利用。纤维素和半纤维素的结构相对简单,而木质素具有复杂的三维无定型结构,相对难以利用,所以目前很多研究与有效的转化利用方法均集中在纤维素和半纤维素上。很多进行生物质物料利用的工厂转化了生物质中的纤维素和半纤维素,剩下的木质素很大部分就被当做废液排出,或当成燃料进行焚烧处理提供能量和蒸汽。这无疑造成了资源的浪费,也带来了潜在的环境污染隐患。因此,尽管木质素高附加值利用的难度很大,人们也在不断对木质素高附加值转化进行研究,采用木质素生产高附加值燃料和精细化学品引起了更多的关注。
并且,由于近年来日益增长的环保需求和能源安全的需要,并基于不与人争粮的原则,木质生物质的转化利用成为生物炼制领域的研究热点,以期减少污染并希望其将来能部分取代日益枯竭的化石资源。木质生物质的转化利用要求充分利用木质生物质的各种成分,木质素就是其中研究的重点。如今,对于木质素的资源化利用也得到了一些成果,主要集中在用作混凝土减水剂、燃料分散剂、农药缓释剂及采油用表面活性剂等领域。然而,这些方面主要是木质素的简单应用,并没有让木质素从根本上得到有效利用。因此,如何有效的对木质素进行解聚使其转化成具有高附加值的化学品,更需要我们的努力。
木质素具有独特的芳香族结构,同时又具有脂肪族结构。从木质素制备高附加值的化学品、燃油替代品和平台化合物等方面的研究有了快速的发展,其中从木质素制备芳香族化合物被认为是最具有前景的方向。从木质素原料得到小分子化合物的方法繁多,大体上有催化热液解聚、高温热解、生物解聚三大类。其中催化热液解聚发展快速,同时也被认为是最具有潜力的解聚方法之一。催化热液解聚根据催化剂种类的不同主要分为催化氧化解聚,催化加氢解聚,酸、碱催化解聚等。
现有技术使用均相酸、碱催化解聚木质素已经具有很广泛的应用,但是不可避免的面临着一些问题,例如后处理复杂、腐蚀反应器、环境污染等。同时,催化氧化解聚带来了产物难以控制、产物种类复杂的问题。采用催化加氢解聚的方式,可选择性地使木质素转化生成芳香族化合物,然而解聚效率低一直是当前木质素解聚所面临的重点问题。
技术实现要素:
有鉴于此,本申请提供一种钌和钨改性的金属固溶体催化剂、其制备方法和解聚木质素的方法,采用本申请所述催化剂催化解聚木质素,可有效提高解聚效率。
本发明提供一种钌和钨改性的金属固溶体催化剂,包括:
锡铝金属固溶体载体;
分散在所述锡铝金属固溶体载体上的钌和钨。
优选地,所述钌和钨改性的金属固溶体催化剂为酸性,具有介孔结构。
优选地,所述钌和钨改性的金属固溶体催化剂中锡和铝的原子比例为0.44:0.53,钌的负载量为1~6wt%,钨的负载量为1~10wt%。
本发明提供一种钌和钨改性的金属固溶体催化剂的制备方法,包括以下步骤:
s1、将锡源和铝源在第一溶剂中混合,通过调节ph值得到沉淀物,将所述沉淀物干燥后进行煅烧,研磨,得到锡铝氧化物;
s2、将所述锡铝氧化物与钨源在第二溶剂中混合,然后依次进行干燥、煅烧,研磨,得到钨改性锡铝氧化物;
s3、将所述钨改性锡铝氧化物与钌源在第三溶剂中混合后,采用还原剂进行还原,得到钌和钨改性的金属固溶体催化剂。
优选地,所述步骤s1具体为:将锡盐和铝盐在水中混合溶解,然后调节ph值为碱性,分离得到沉淀物,将所述沉淀物洗涤至中性,干燥,煅烧,研磨,得到锡铝氧化物;所述煅烧的温度优选为400~650℃。
优选地,所述步骤s2具体为:将所述锡铝氧化物与钨盐在水中混合溶解,然后干燥,煅烧,研磨,得到钨改性锡铝氧化物;所述干燥的温度优选为60~80℃;所述煅烧的温度优选为400~650℃。
优选地,所述步骤s3具体为:将所述钨改性锡铝氧化物与钌盐在第三溶剂中混合溶解,加入硼氢化钠还原,分离得到固相,干燥得到最终的金属固溶体催化剂;所述第三溶剂优选为醇水混合溶液。
本发明提供一种解聚木质素的方法,包括以下步骤:
在催化剂存在的条件下,将木质素在有机溶剂中进行加氢解聚反应,得到含有小分子化合物的解聚产物;
所述催化剂为前文所述钌和钨改性的金属固溶体催化剂。
优选地,所述木质素和催化剂的质量比为(1~20):1;所述有机溶剂为二氧六环和甲醇的混合溶液,所述二氧六环和甲醇的体积比为(1~15):1。
优选地,所述加氢解聚反应的温度为150~400℃,压力为1~5mpa,时间为1~24h。
与现有技术相比,本发明提供一种钌和钨改性的金属固溶体催化剂,其以锡铝金属固溶体为载体,并且采用钨和钌对催化剂进行改性。所述钌和钨改性的金属固溶体催化剂可采用如下方法制备:第一步将锡源、铝源在溶剂中搅拌混合,调节ph进行沉淀,然后过滤,洗涤,干燥,煅烧,研磨;第二步将得到的锡铝氧化物与钨源在溶剂中搅拌混合,然后干燥、煅烧,研磨;第三步将得到的钨改性锡铝氧化物与钌源在溶剂中还原,然后过滤、干燥,得到最终的催化剂。本发明所述催化剂主要使用锡和铝复配的金属固溶体作为载体,使用钨改性增强了催化剂的酸性;钌改性增强了催化剂的效果。因此,本发明所述钌和钨改性的金属固溶体催化剂能够高效解聚木质素。
应用本发明所述钌和钨改性的金属固溶体催化剂催化解聚木质素,可以降低催化剂对于反应器的伤害,解聚率高。同时,所得解聚产物中芳香族化合物产率和石油醚萃取的小分子化合物产率高,分离提纯简单。本发明的解聚产物和催化剂的分离简单,对环境友好。
附图说明
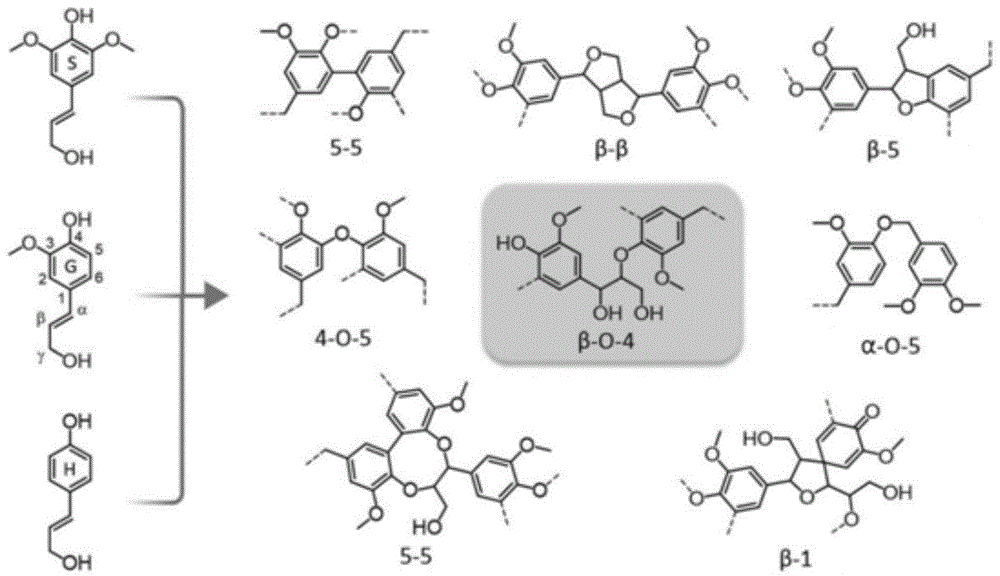
图1为本发明一些实施例中木质素的典型键合结构图;
图2为本发明实施例1、比较例1和比较例2所述催化剂的xrd结果;
图3为本发明实施例1所述催化剂的sem照片(标尺2μm);
图4为本发明实施例1所述催化剂的sem照片(标尺1μm)。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种钌和钨改性的金属固溶体催化剂,包括:
锡铝金属固溶体载体;
分散在所述锡铝金属固溶体载体上的钌和钨。
本发明提供的催化剂对木质素有高效的解聚效果,并且易于与解聚产物分离,对环境污染小。
本发明所述的催化剂为钌和钨改性的锡铝金属固溶体催化剂,其包括锡铝金属固溶体载体。在本发明中,主要采用锡(sn)和铝(al)复配的金属固溶体作为载体。固溶体是指溶质原子溶入金属溶剂晶格中而仍保持溶剂类型的合金相;在本发明实施例中,通过x射线衍射(xrd)分析,al很好地分散到sn的晶格当中,形成了金属固溶体,可表示为sn-alox。具体地,所述钌和钨改性的金属固溶体催化剂中,锡和铝的原子比例为0.44:0.53。
在组分上,本发明所述钌和钨改性的金属固溶体催化剂还含有钌(ru)和钨(w)。在本发明实施例中,w和ru均匀地分散在所述锡铝金属固溶体载体上。其中,使用钨改性增强了催化剂的酸性;使用钌改性增强了催化剂的效果。具体地,所述钌和钨改性的金属固溶体催化剂中,钌的负载量可为1~6wt%,优选2~5wt%;钨的负载量可为1~10wt%,优选10wt%。
在本发明的实施例中,所述钌和钨改性的金属固溶体催化剂整体为酸性。并且,该催化剂具有良好的比表面积,同时具有一定的介孔结构。其中,所述催化剂的bet比表面积例如在145m2/g以上。根据扫描电子显微镜(sem)等分析,所述的介孔结构可由棒状的ru-w/sn-alox晶体堆叠而成。
本发明所述钌和钨改性的锡铝金属固溶体催化剂为多元复配金属催化剂,各组元综合作用下,本发明该金属固溶体催化剂相对于目前一般使用的催化剂有明显更高的解聚效果,以及更高的产物产率等。
相应地,本发明实施例提供了一种钌和钨改性的金属固溶体催化剂的制备方法,包括以下步骤:
第一步、将锡源和铝源在第一溶剂中混合,通过调节ph值得到沉淀物,将所述沉淀物干燥后进行煅烧,研磨,得到锡铝氧化物;
第二步、将所述锡铝氧化物与钨源在第二溶剂中混合,然后依次进行干燥、煅烧,研磨,得到钨改性锡铝氧化物;
第三步、将所述钨改性锡铝氧化物与钌源在第三溶剂中混合后,采用还原剂进行还原,得到钌和钨改性的金属固溶体催化剂。
采用本申请实施例制备的催化剂催化解聚木质素,解聚效率高,解聚产物分离简单。
本发明实施例第一步将锡源、铝源在溶剂中优选搅拌混合,然后调节混合溶液ph值进行沉淀,可过滤得到沉淀物,再将其洗涤,干燥,煅烧,研磨,得到锡铝氧化物。
其中,本发明对于锡源、铝源不进行特殊限定,本领域技术人员熟知的可以提供锡原子、铝原子的化合物即可。所述的锡源、铝源一般为对应金属的盐类物质,优选可以为氯化锡、硝酸锡、氯化铝、硝酸铝,更优选为氯化锡、硝酸铝。在本发明中,上述第一步中锡源、铝源对应的盐类物质质量比优选为5:(3~7)。该步骤所用溶剂记为第一溶剂,后续步骤以此类推;本发明所述第一溶剂优选水。本发明对于所述水的来源不进行限定,本领域技术人员熟知的即可,实验室通常采用蒸馏水。
本发明实施例所述的第一个步骤具体为:将锡盐和铝盐加入水中搅拌混合,溶解得到混合溶液,然后使用碱性物质调节ph值为碱性,再过滤分离得到沉淀物,优选将所述沉淀物洗涤至中性(例如ph值为7~8),之后干燥,煅烧,研磨得到锡铝氧化物。其中,本发明对于所述搅拌的具体方式不限定,所述搅拌的时间优选为1~5h;更优选为2~4h。所述搅拌的温度优选为20~80℃;更优选为30~70℃。所述搅拌速度优选为500~700rpm;更优选为600~700rpm。本发明对于调节ph进行沉淀的具体操作不进行限定,本领域技术人员熟知的即可。本发明实施例ph值调节优选使用的碱性物质为氢氧化钠、氢氧化钾和氨水中的一种或多种;优选调节ph值至10~12以得到沉淀物。
本发明实施例用水洗涤所述沉淀物后干燥,最后煅烧,研磨即得所述的氧化物。本发明对于所述干燥的具体方式不进行限定,本领域技术人员熟知的即可。所述干燥的温度优选为60~80℃,更优选为65~75℃;所述干燥的时间优选为6h~24h,更优选为8~22h,最优选为10~20h。本发明对于所述煅烧、研磨的具体方式不进行限定,本领域技术人员熟知的即可。所述煅烧的温度优选为400~650℃,更优选为450~630℃,最优选为500~600℃;所述煅烧的时间优选为3h~10h,更优选为3~8h,最优选为3~6h。
得到锡铝氧化物后,本发明实施例第二个步骤是将其与钨源在第二溶剂中搅拌混合,然后干燥,煅烧,研磨,得到钨改性锡铝氧化物。
其中,本发明对于钨源不进行限定,本领域技术人员熟知的可以提供钨原子的化合物即可;具体可为钨源对应的金属盐,优选为磷钨酸或钨酸铵。在本发明中,上述第一步所得氧化物与第二步新增的氧化物的质量比可为20:(1~5)。在本发明中,上述第二步中所述第二溶剂优选为水。本发明对于所述水的来源不进行限定,本领域技术人员熟知的即可。本发明对该步骤中所述搅拌、干燥和煅烧等的具体方式也不进行特殊限定,可与第一步中相应操作相同。例如,所述干燥的温度优选为60~80℃;所述煅烧的温度优选为400~650℃。
本发明实施例通过上述处理使钨改性催化剂,得到钨改性锡铝氧化物。本发明实施例第三步将上述制备出的特定氧化物与钌源在第三溶剂中搅拌混合,溶解后加入还原剂进行还原,然后过滤,干燥,得到最终的金属固溶体催化剂。
其中,本发明对于钌源不进行限定,本领域技术人员熟知的可以提供钌原子的化合物即可;具体可为钌源对应的金属盐,优选为氯化钌或硝酸钌。在本发明中,上述第二步得到的氧化物与第三步负载的金属的质量比可为20:(1~5)。在本发明中,上述第三步中所述第三溶剂优选为醇水混合溶液,更优选为体积比为1:1混合的水和乙醇。本发明对于所述水和乙醇的来源不进行限定,本领域技术人员熟知的即可。
本发明实施例将所述钨改性锡铝氧化物加入至第三溶剂后,加入钌盐搅拌混合,溶解,加入硼氢化钠还原,可过滤分离得到固相,用水洗涤后干燥,得到最终的催化剂。在本发明的实施例中,所述还原过程中优选硼氢化钠过量,在室温下,还原时间可为5~10h,优选8h。所述干燥的温度优选为60~80℃,更优选为65~75℃;所述干燥的时间优选为6h~24h,更优选为8~22h,最优选为10~20h。
本发明实施例最终得到的催化剂为前文所述钌和钨改性的锡铝金属固溶体催化剂,可记为ru-w/sn-alox。本发明所述钌和钨改性的锡铝金属固溶体催化剂主要应用于木质素催化解聚,可高效解聚木质素。
本发明实施例还提供了一种解聚木质素的方法,包括以下步骤:
在催化剂存在的条件下,将木质素在有机溶剂中进行加氢解聚反应,得到含有小分子化合物的解聚产物;所述催化剂为前文所述钌和钨改性的金属固溶体催化剂。
本发明催化解聚木质素的方法具有解聚效率高,解聚产物分离简单,环境友好等特点。
本发明实施例采用一定量的前文所述钌和钨改性的金属固溶体催化剂,催化解聚木质素。所述的木质素是自然界中第二丰富的天然高分子,其与纤维素、半纤维素一起组成植物的主要结构。木质素是一种广泛存在于植物体中的无定形的、分子结构中含有氧代苯丙醇或其衍生物结构单元的芳香性高聚物。木质素是由聚合的芳香醇构成的一类物质,存在于木质组织中,主要作用是通过形成交织网来硬化细胞壁。木质素主要位于纤维素纤维之间,起抗压作用。在木本植物中,木质素占25%,是世界上第二位最丰富的有机物。木质素的来源有很多,本发明对此不进行限定,可以为:造纸木质素、玉米芯水解木质素、从桉树中稀酸水解木质素、从松木中有机溶剂法得到的木质素,从杏壳中有机溶剂法获取木质素。在本发明的一些实施例中,所述木质素为松木硫酸盐木质素。
本发明实施例采用上述催化剂催化解聚木质素,具体为:采用催化剂、在有机溶剂中溶解,在反应釜中充入氢气将木质素解聚,得到解聚后的产物。在本发明中,所述木质素和催化剂的质量比优选为(1~20):1,更优选为(2~18):1,最优选为(3~16):1。在本发明中,所述催化解聚的有机溶剂优选为二氧六环和甲醇的混合溶液;所述二氧六环和甲醇的体积比优选为(1~15):1,更优选为(2~14):1,最优选为(3~13):1。所述溶解优选为超声溶解;本发明对于所述反应容器不进行限定,本领域技术人员熟知的即可,可以为反应釜。反应中优选充入氢气,密封。
本发明所述加氢解聚反应的温度优选为150~400℃;更优选为170~400℃。所述加氢解聚反应的压力优选为1~5mpa;更优选为2~4mpa;所述的解聚时间优选为1h~36h;更优选为6~30h;最优选为12~24h。本发明实施例通过加热升温至特定反应温度;所述升温速度优选为1~10℃/min。
解聚反应结束后,本发明实施例优选冷却反应体系。所述冷却优选冷却至室温;本发明对于所述冷却的方式不进行限定。冷却后,收集反应溶液,通过过滤分离出催化剂和残渣,得到滤液,进入气相测定部分为芳香族化合物。本发明实施例所述芳香族化合物包括:苯酚、2-甲氧基酚、2-甲氧基苯酚、邻二甲氧基苯、4-甲基愈创木酚、4-乙基愈创木酚、丁香酚、3,4-二甲氧基甲苯、4-丙基愈创木酚、香草醛、异丁香酚、香草乙酮、4-羟基-3甲氧基苯丙酮、2,6-二叔丁基-4-甲基苯酚和高香草酸。
在本发明的实施例中,所得滤液浓缩后用丙酮溶解,而后用石油醚萃取为小分子组分;所述浓缩的方式优选为旋蒸。
本发明采用上述特定的方法制备的钌和钨改性的锡铝金属固溶体催化剂,其对木质素的解聚率高,对反应器伤害小,并且之后的催化剂分离简单。同时,本发明所述的钌和钨改性的锡铝金属固溶体催化剂的稳定性好,在320℃仍然稳定性良好。
为了进一步理解本申请,下面结合实施例对本申请提供的钌和钨改性的金属固溶体催化剂、其制备方法和解聚木质素的方法进行具体地描述。但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
以下实施例中,木质素来源为购买自美德维实伟克公司(meadwestvaco)无锡公司,indulinat为松木硫酸盐木质素,其结构性能参数如下表和图1,图1为木质素的一些典型键合结构,可以得到表2里的一些结构β和4,5都是碳的位置。此外,以下实施例所涉及的搅拌速度均为600rpm。
表1.indulinat木质素的元素含量和甲氧基含量
表2.indulinat木质素的结构
实施例1
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与0.8208g氯化钌搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,室温下搅拌还原8h(以下实施例还原参数相同),过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂ru-w/sn-alox。
该催化剂中ru的负载量是5wt%,w的负载量是10wt%,sn和al原子比例为0.44:0.53。
比较例1
将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,得到锡铝催化剂sn-alox。
比较例2
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨得到钨改性锡铝催化剂w/sn-alox。
图2是实施例1、比较例1和比较例2的xrd表征图,图中的峰均是四方晶型的sno2的特征峰,没有al、w和ru相关的峰出现,这说明实施例1的催化剂中w和ru均匀的分散在sn-alox的表面。同时,一般来说通过al(oh)3煅烧制备成的al2o3,在煅烧温度为450度以上就会形成晶体,但是在al含量比较大的情况下,没有al相关的晶体衍射峰出现,这说明al很好地分散到sn的晶格当中,形成了金属固溶体。
表3表示了实施例1、比较例1和比较例2的氨气升温脱附程序结果,一般来说,氨气脱附峰的温度越高,说明对应酸性活性位的酸性越强,脱附的量越大,说明对应活性位的数量约多。从表中可以看出,w改性之后出现了一个酸性很强的活性位(3号峰),ru继续改性之后虽然这个酸性很强的3号峰被掩盖了,但是实施例1所述催化剂整体的酸性还是继续增强的。
表3氨气脱附结果
a氨气脱附温度。b氨气脱附量。
表4是通过bet表征得到实施例1、比较例1和比较例2的催化剂的结构性质,从表中可以看出,通过实施例1这种方法制备的催化剂具有良好的比表面积,同时具有一定的介孔结构。
图3-4是实施例1所述催化剂的sem结果,从图中可以看出,这介孔结构是由棒状的ru-w/sn-alox晶体堆叠而成。
表4催化剂结构性质
adeterminedfromtheamountadsorbedatp/p0=0.95.
比较例3
将0.5g木质素溶解在体积比为5:1的二氧六环和甲醇混合溶液中,超声后加入50ml高压反应釜中。同时向该反应釜中加入0.1g比较例1制备的催化剂,充入2mpa氢气,密封之后搅拌速率调整为900rpm。用1h升温至300℃,反应1h,反应结束后迅速冷却至室温。收集反应后溶液,抽滤分离出催化剂和残渣,在用pes针头过滤,得到滤液。
加入标物,稀释之后,用gc检测芳香族化合物的含量。检测条件为:shimadzugc-2010,wondacap5column,检测器为fid,炉温为50℃维持3min,以10℃/min升温到250℃,检测器的温度维持在280℃,氦气作为载气。
对上述滤液进行旋转蒸发,最终得到的产物用1ml丙酮溶解,加入200ml石油醚萃取石油醚萃取物。结果表明,该条件下芳香族化合物的收率为5.97%,石油醚萃取物收率为14.93%。
比较例4
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g比较例2制备的催化剂。结果表明,芳香族化合物收率为7.81%;石油醚萃取物收率为21.1%。
比较例5
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g比较例2制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率为14.78%;石油醚萃取物收率为36.8%。
实施例2
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g实施例1制备的催化剂。结果表明,芳香族化合物收率为7.48%;石油醚萃取物收率为20.19%。
实施例3
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g实施例1制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率分别为15.69%;石油醚萃取物收率分别为59.86%。
具体反应条件及结果参见表5。从表5的结果可以看出,加入制备催化剂的几步措施均有其改进效率之处和改进的必要性,对于反应效率具有明显的提升。在其他条件不变的情况下,适当增加反应时间有明显的好处。
表5本发明实施例2~3和比较例3~5所述的反应条件及结果
实施例4~6
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g实施例1制备的催化剂,反应温度分别为270℃、290℃和310℃。结果表明,芳香族化合物收率分别为7.49%、10.39%、17.22%;石油醚萃取物收率分别为20.47%、25.46%、53.58%。
具体反应条件及结果如表6所示。由表6的结果可以看出,在其他条件不变的情况下,增加反应温度到300℃可以很大程度上增加解聚的效果,在300℃以上再增加温度的效果提升不大。
表6本发明实施例3~6所述的反应条件及结果
比较例6
具体反应过程和检测方法与比较例3相同,只是不投入催化剂,反应时间为1h。结果表明,芳香族化合物收率为4.13%;石油醚萃取物收率为10.11%;总液体产物收率为56.06%。
比较例7
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1ghzsm-5沸石固体酸催化剂,反应时间为12h。结果表明,芳香族化合物收率为12.18%;石油醚萃取物收率为31.76%;总液体产物收率为67.81%。
比较例8
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1gh-βeta沸石固体酸催化剂,反应时间为12h。结果表明,芳香族化合物收率为11.22%;石油醚萃取物收率为35.21%;总液体产物收率为73.84%。
比较例9
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1gru/c催化剂(ru负载量为5wt%的商业ru/c催化剂)。结果表明,芳香族化合物收率为5.85%;石油醚萃取物收率为12.91%;总液体产物收率为75.32%。
比较例10
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1gru/c催化剂,反应时间为12h。结果表明,芳香族化合物收率为14.33%;石油醚萃取物收率为46.99%;总液体产物收率为95.77%。
具体反应条件及结果如表7所示。由表7的结果可以看出,本发明制备的多元复配金属催化剂相对于目前一般使用的催化剂有明显更高的解聚效果,和更高的产物产率。
表7本发明实施例2~3和比较例6~10所述的反应条件及结果
比较例11
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与0.8660g二水合硝酸钯搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂。
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g本比较例中制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率为13.79%;石油醚萃取物收率为56.19%;总液体产物收率为93.39%。
比较例12
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与0.6544g硝酸铂搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂。
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g本比较例中制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率为14.43%;石油醚萃取物收率为55.97%;总液体产物收率为94.75%。
比较例13
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与1.9819g六水合硝酸镍搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂。
具体反应过程和检测方法与比较例3相同,只是投入的催化剂0.1g本比较例中制备得到的催化剂,反应时间为12h。结果表明,芳香族化合物收率为13.69%;石油醚萃取物收率为48.86%;总液体产物收率为85.05%。
具体反应条件及结果如表8所示;从表8的结果可以看出,钌有相对更好的催化效果。
表8本发明实施例3和比较例11~13所述的反应条件及结果
实施例7
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与0.563g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与0.8208g氯化钌搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂ru-w/sn-alox。
实施例8
第一步、将15.381g的氯化锡和19.732g的硝酸铝搅拌溶解于50ml水中,使用氢氧化钠溶液将ph调节至12,沉淀,过滤,用蒸馏水洗涤沉淀物至ph值在7~8之间,室温搅拌3h。将洗涤后的固相于80℃干燥10h,550℃煅烧3h,研磨。
第二步、将8g第一步得到的氧化物与1.126g磷钨酸在50ml水中混合,80℃搅拌3h,之后80℃干燥10h,500℃煅烧3h,研磨。
第三步、将8g第二步得到的氧化物与0.4104g氯化钌搅拌混合在50ml溶剂中(体积比为1:1的水和乙醇混合溶液),投入16g过量的硼氢化钠还原,过滤,用蒸馏水洗涤固相,将洗涤后的固相于80℃干燥10h,得到最终的催化剂ru-w/sn-alox。
实施例9
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g实施例7制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率为13.17%;石油醚萃取物收率为55.24%。
实施例10
具体反应过程和检测方法与比较例3相同,只是投入的催化剂为0.1g实施例8制备的催化剂,反应时间为12h。结果表明,芳香族化合物收率为14.42%;石油醚萃取物收率为52.61%。
由以上实施例可知,本发明所述催化剂主要使用锡和铝复配的金属固溶体作为载体,使用钨改性增强了催化剂的酸性;钌改性增强了催化剂的效果。本发明所述钌和钨改性的金属固溶体催化剂能够高效解聚木质素;本发明解聚木质素的方法可以降低催化剂对于反应器的伤害,解聚率高。同时,所得解聚产物中芳香族化合物产率和石油醚萃取的小分子化合物产率高,分离提纯简单。本发明的解聚产物和催化剂的分离简单,对环境友好。
以上所述仅是本发明的优选实施方式,应当指出,对于使本技术领域的专业技术人员,在不脱离本发明技术原理的前提下,是能够实现对这些实施例的多种修改的,而这些修改也应视为本发明应该保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!