一种芳香化合物催化燃烧催化剂及其制备和应用的制作方法
[0001]
本发明属于环境催化领域,涉及一种芳香化合物催化燃烧催化剂。
背景技术:
[0002]
挥发性有机化合物(vocs,volatile organic compounds)是主要的大气污染物,主要来源于燃料燃烧、工业制造和溶剂排放,对大气环境和人体健康带来严重危害。芳香化合物,如甲苯,是一种主要的vocs污染物。作为一种常见的溶剂,甲苯被广泛应用于装饰材料和油漆涂料等化学品制造业。如何将各行业产生的甲苯废气有效转化或去除,具有重要意义。
[0003]
常见的空气中甲苯的去除方法有:吸附法、光催化降解法、等离子体降解法、无氧化燃烧法、催化氧化(燃烧)法等。催化氧化法可在较低温度下利用催化剂的催化氧化性将甲苯转化为二氧化碳和水,具有操作简单、能耗低、效率髙、无二次污染、适用范围广等优点,是目前受到世界公认的最有效的方式。
[0004]
甲苯催化燃烧的催化剂,主要有三种:一是负载型贵金属催化剂,以pt、pd、au、ru等贵金属为代表;二是过渡金属氧化物催化剂,常见的有v2o5、mnox、co3o4、ceo、cuo、fe2o3等:三是复合氧化物催化剂,主要是钙钛矿型复合氧化物和尖晶石性复合氧化物。过渡金属氧化物和复合金属氧化物,价格相对便宜,但存在起燃温度高、催化活性低的问题。相对而言,负载型贵金属催化剂具有更好的催化活性、更低的起燃温度和完全转化温度,在催化氧化vocs领域得到广泛研究。
[0005]
用于甲苯催化燃烧的负载型贵金属催化剂,研究的重点是提高催化剂活性。一般来说,催化剂的活性越高,甲苯的起燃温度和完全转化温度越低。目前的研究方向主要有两个。一个是合金化,即通过两种金属间的相互作用,调变活性金属的表面电子性质,提高催化活性;另一个是增大金属的分散度,即通过改变载体类型和优化负载方法,提高金属的分散度,提高催化活性。例如,专利cn101733165a、cn201110064279和cn201610223024.2报道了以堇青石和氧化铝作为载体制备pd和pt整体式催化剂,在温度为182-250℃时,对甲苯的转化率达99%。而且,pd和pt相比,pd具有成本低的优点,是工业生产与应用的最佳活性组分之一。cn 109225319以sapo-34分子筛为载体制备了pt/sapo-34分子筛催化剂,140℃-250℃下,甲苯催化燃烧。cn106362736报道了一种低负载量0.05%以下pd@pt/al2o3核壳结构催化剂,使得甲苯在约150℃下起燃,220℃完全转化。cn108906039a报道了一种低负载量的au@pt/al2o3核壳结构催化剂,在210℃将甲苯完全氧化。
[0006]
综上,现有的甲苯催化氧化催化剂还存在着起燃温度(t
10
)和完全转化温度(t
90
)温度较高的不足。
技术实现要素:
[0007]
针对现有芳香化合物催化燃烧催化剂存在催化性能不理想,起燃温度和完全转化温度高的技术不足,本发明第一目的在于,提供一种芳香化合物催化燃烧催化剂(本发明也
简称为催化剂),旨在提供一种具有优异催化活性,较低起燃温度和完全转化温度的全新催化剂。
[0008]
本发明第二目的在于,提供所述的催化剂的制备方法。
[0009]
本发明第三目的在于,提供所述的催化剂在芳香化合物催化燃烧中的应用。
[0010]
一种芳香化合物催化燃烧催化剂,包括具有细胞壁阵列结构的木材基底,细胞壁的骨架表面原位复合有碳膜以及弥散分布的氧化石墨烯包覆的贵金属纳米颗粒。
[0011]
本发明提供了一种全新结构的催化剂,其创新地利用木材原生的细胞壁结构为担载基底,且在细胞壁的阵列骨架(细胞壁框架)表面原位复合有碳膜以及氧化石墨烯包覆的贵金属纳米颗粒(标记为go@m)。研究发现,本发明所述的催化剂,基于成分以及形貌结构的协同,能够意外地改善其在芳香化合物的催化燃烧过程的催化活性,降低起燃温度和完全转化温度,此外,还具有优异的结构稳定性和催化循环稳定性。
[0012]
本发明中,所述的木材原生(天然)细胞壁生物质框架结构及其表面的碳膜包覆结构和go@m均匀弥散分布结构特性是赋予所述催化剂在芳香化合物中优异催化性能的关键。
[0013]
本发明研究发现,对木材细胞壁框架结构进行调控,有助于进一步和所述的表面原位碳膜和go@m结构特性协同,有助于进一步改善其在芳香化合物催化燃烧过程中的催化活性。
[0014]
作为优选,所述的木材具有40~60μm的纵向直通孔道,5~15μm的横向纹孔。
[0015]
优选地,所述的木材为阔叶林木,进一步优选为杨木、杉木、榉木中的至少一种;最优选为杨木。本发明意外地发现,采用杨木作为原位生物质框架,配合所述的表面原位碳膜以及go@m结构特性,有助于进一步改善催化剂的催化活性。
[0016]
本发明中,控制贵金属的成分,有助于进一步协同改善所述催化剂的催化活性。
[0017]
作为优选,所述的贵金属为钯、铂或钯铂合金;优选为钯。本发明研究发现,所述的钯有助于进一步和所述的结构协同,进一步改善催化性能。
[0018]
所述的贵金属纳米颗粒的粒径为1-5nm。
[0019]
本发明中,所述的贵金属由氧化石墨烯膜包覆,所述的氧化石墨烯的厚度不高于5nm。
[0020]
本发明中,所述的碳膜由细胞壁表面原位转化得到,其厚度不高于5nm。
[0021]
作为优选,贵金属的负载量为0.01%wt-0.5%wt。
[0022]
本发明还提供了一种所述的芳香化合物催化燃烧催化剂的制备方法,包括以下步骤:
[0023]
步骤(1):将维持有原有细胞壁阵列结构的木材置于溶解有贵金属源的溶液中;进行浸渍;随后干燥,得到负载有贵金属源的木材;
[0024]
步骤(2):将负载有贵金属源的木材进行还原处理,将贵金属源还原成单质,得到负载有贵金属单质的木材;
[0025]
步骤(3):将负载有贵金属单质的木材在保护气氛、200-350℃的温度下对木材细胞壁骨架进行表面原位碳化,制得所述的催化剂。
[0026]
本发明研究发现,成功维持木材细胞壁原生生物质框架结构、避免结构坍塌、控制碳膜原位转化程度以及go@m的原位复合结构是成功构建所述的材料,改善其催化燃烧性能的关键。为此,本发明创新地发现,采用木材作为基底,预先在其细胞壁结构中还原形成贵
金属纳米颗粒,随后再在所要求的温度下进行细胞壁表面处理,从而实现细胞壁表面的原位镀碳膜,并避免细胞壁结构不被碳化,不仅如此,还实现所述的贵金属颗粒表面的原位氧化石墨烯包覆。本发明研究发现,基于所述的原料、浸渍-还原以及特殊温度下的可控表面处理工艺,可以意外地构建所述全新结构的催化剂,并改善制得的催化剂的催化性能。
[0027]
本发明中,所述的木材为段状的原生木材。所述的段状结构区别于常规的粉碎原料,旨在从原料的基础上提供木材原生的细胞壁框架结构。
[0028]
本发明中,所述的木材可以是直接取自原生木材段状(块体)结构的树干组织,例如,可以是取自木材的柱体结构树干组织(例如为圆柱体)。所述的柱体长度方向优选为所述的木材的高度方向。
[0029]
本发明中,所述的贵金属源为贵金属元素的水溶性盐,优选为贵金属元素的氯化盐、硝酸盐、硝酸盐、醋酸盐、乙酰丙酮盐中的至少一种;
[0030]
优选地,所述的浸渍过程的温度为0-40℃,进一步优选为20-30℃;
[0031]
优选地,浸渍时间为20-90天,进一步优选30-40天。
[0032]
步骤(2)的还原为液相还原或者气相还原。
[0033]
所述的液相还原指将步骤(1)处理后的木材置于含有还原剂的溶液中,利用还原剂将贵金属源还原成单质。
[0034]
所述的气相还原指:将所述的负载有贵金属源的木材置于还原性气氛中剂型气固还原。
[0035]
作为优选,所述的还原步骤为气相还原。
[0036]
优选地,所述的还原性气氛为含有氢气的气氛。
[0037]
优选地,气相还原过程中的温度为50-200℃,优选100-150℃的温度下进行还原。
[0038]
本发明中,步骤(3)的表面处理的温度控制是成功制备所述的催化剂的关键之一。研究发现,基于所述的温度的控制,从而实现细胞壁框架结构的表面可控碳膜构建,并避免整体结构碳化以及结构原生结构坍塌,如此可以意外地改善材料的在芳香化合物中的催化性能。
[0039]
作为优选,表面原位碳化过程的温度为200~350℃;进一步优选为250~320℃。
[0040]
优选地,表面原位碳化的时间为0.5-5小时,优选1-3小时。
[0041]
本发明还提供了所述的芳香化合物催化燃烧催化剂的应用,将其用作催化剂,催化芳香化合物氧化降解。
[0042]
优选地,芳香化合物为苯、烷烃基取代苯、卤代苯中的至少一种;进一步优选为甲苯、二甲苯中的至少一种。
[0043]
优选的应用,催化过程的温度大于或等于75℃;优选为130~150℃。
[0044]
有益效果
[0045]
1、本发明提供了一种全新结构的催化剂,且发现其在芳香化合物催化燃烧方面具有意料不到的技术效果,具有更低的起燃温度和完全转化温度,具有优异的结构和循环稳定性。
[0046]
例如,与现有技术相比,本发明所述的催化剂具有催化活性高(甲苯起燃温度75-100℃,完全转化温度135-210℃)、载体成本低且环境友好(经济阔叶林木)、制备方法简单以及稳定性好的优点。
[0047]
2、本发明还提供了所述的催化剂的制备方法,其基于所述的原料的使用,并配合所述的浸渍-还原-表面处理工艺的协同,能够成功维持木材原生结构,并在细胞壁的表面原位形成碳膜以及所述的氧化石墨烯包覆的贵金属颗粒。基于本发明的制备工艺和条件的控制,能够制备一种在芳香化合物催化燃烧方面具有优异性能的催化剂材料。
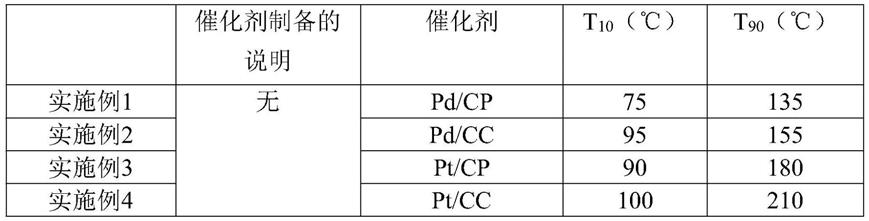
附图说明
[0048]
图1为实施例1制得的pd/cp催化剂高分辨透射电镜(tem)图
[0049]
图2杨木的tem图
[0050]
图3pd/cp和pd/np催化剂的x射线衍射谱(xrd)
[0051]
从图1可以看出,本发明的催化剂,原位低温炭化处理后,框架结构的表面有碳膜,弥散分布的钯纳米颗粒表面有明显的go薄膜。
[0052]
从图2可以看出,原生杨木低温炭化后保留完好的纵向直通孔道结构,以及横向的互通孔道。
[0053]
从图3可以看出,本发明的催化剂碳化结构,都保留有纤维素的峰。说明细胞壁结构的存在。
具体实施方案
[0054]
以下结合具体的实施例和对比例,进一步详细描述本发明的技术方案,但所述实施例不限制本发明的保护范围。需要说明的是,为了保证催化剂的可比性,钯的实际负载量控制在0.23-0.25wt%之间,浮动(偏差)控制在2%之内。
[0055]
实施例1
[0056]
将原生白杨木(河南焦作产,去皮,切成直径(宽)1厘米、长度5厘米的圆柱)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温,进行表面处理2小时,降温冷却,得到钯质量百分数为0.23wt%的pd/表面碳化杨木催化剂,记为pd/cp。
[0057]
实施例2
[0058]
和实施例1相比,区别仅在于,木材种类不同,具体为:
[0059]
将原生巴尔杉木(南美洲产,去皮,切成直径1厘米、长度5厘米的圆柱)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温进行表面处理2小时,降温冷却,得到钯质量百分数为0.25wt%的pd/表面碳化杉木催化剂,记为pd/cc。
[0060]
实施例3
[0061]
和实施例1相比,区别仅在于,贵金属源为pt源,具体为:
[0062]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯铂酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到铂质量百分数为0.23wt%的pt/表面碳化杨木催化剂,记为pt/cp。
[0063]
实施例4
[0064]
和实施例2相比,区别仅在于,贵金属源为pt源,具体为:
[0065]
将原生巴尔杉木(同实施例2)在浓度为0.022mol/l的氯铂酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到铂质量百分数为0.25wt%的pt/表面碳化杉木催化剂,记为pt/cc。
[0066]
实施例5
[0067]
和实施例1相比,区别在于,表面处理过程的温度为200℃,具体为:
[0068]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h,降温冷却。再于氮气气氛下,200℃保温处理2小时,得到钯质量百分数为0.23wt%的pd/低温表面碳化杨木催化剂,记为pd/lcp。
[0069]
实施例6
[0070]
和实施例1相比,区别在于,还原过程采用液相还原手段,具体为:
[0071]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用浓度为0.1mol/l的硼氢化钠溶液室温静置浸泡,还原2h。最后在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到液相还原的钯质量百分数为0.23wt%的pd/表面碳化杨木催化剂,记为pd/rcp。
[0072]
对比例1
[0073]
和实施例1相比,区别仅在于,未进行表面碳化处理,具体为:
[0074]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h,降温冷却,得到钯质量百分数为0.23wt%的pd/未碳化杨木催化剂,记为pd/np。
[0075]
对比例2
[0076]
和实施例2相比,区别仅在于,未进行表面碳化处理,具体为:
[0077]
将原生巴尔杉木(同实施例2)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h,降温冷却,得到钯质量百分数为0.23wt%的pd/未碳化杉木催化剂,记为pd/np。
[0078]
对比例3
[0079]
和实施例1相比,区别仅在于,表面碳化处理处理的温度为500度,具体为:
[0080]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h,降温冷却。再于氮气气氛下,500℃保温处理2小时,得到钯质量百分数为0.23wt%的pd/高温碳化杨木催化剂,记为pd/hcp。
[0081]
对比例4
[0082]
和实施例1相比,区别主要在于,浸渍、还原同步进行,具体为:
[0083]
将原生白杨木(同实施例1)在浓度为0.022mol/l的氯钯酸水溶液中,采用原位浸渍-还原的方法,即在浸渍的同时缓慢滴加硼氢化钠进行还原3小时。取出在阴凉处晾干后,在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到钯质量百分数为0.24wt%的催化剂,记为pd/cp(m1)。
[0084]
对比例5
[0085]
采用“碳化-浸渍-还原工艺”制备0.238wt%的pd/碳化杨木催化剂,具体为:
[0086]
将原生白杨木(同实施例1),在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到碳化过的白杨木。然后在浓度为0.022mol/l的氯钯酸水溶液中,浸渍30天。取出在阴凉处晾干,用流量为30ml/min的氢气180℃还原2h。降温取出后,得到钯质量百分数为0.238wt%的催化剂,记为pd/cp(m2)。
[0087]
对比例6
[0088]
和实施例1相比,区别在于,未保留木材原生细胞壁阵列,具体为:
[0089]
将原生白杨木(同实施例1)用球磨机磨碎,取40-60目的粉末,在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到钯质量百分数为0.243wt%的催化剂,记为pd/cp(粉)。
[0090]
对比例7
[0091]
和实施例1相比,区别在于,采用竹子替换本发明所述的木材,具体为:
[0092]
将原生竹子(湖南益阳产,去皮,切成直径1厘米、长度5厘米的圆柱),在浓度为0.022mol/l的氯钯酸水溶液中,室温浸渍30天。取出在阴凉处晾干后,用流量为30ml/min的氢气180℃还原2h。最后在氮气气氛下5℃/min升温至300℃,保温处理2小时,降温冷却,得到钯质量百分数为0.232wt%的催化剂,记为pd/bamboo催化剂。
[0093]
所有不同实施例、对比例与文献催化剂的甲苯催化燃烧的性能对比如表1所示。
[0094]
甲苯催化燃烧性能的测定
[0095]
催化剂对甲苯的低温催化燃烧性能测试条件如下:将1g催化剂装填于管式反应器中,然后通过氮气鼓泡带甲苯,并与空气混合后进入反应器。进气流量为30~100ml/min,空速6000h-1
。反应气中甲苯的浓度为5000ppm,其他气体成分及组成如表1所示。缓慢升温,出口气体的组成通过气相色谱仪在线监测,并计算甲苯的转化率。将甲苯转化率为10%和90%时的温度,分别记为t
10
和t
90
,分别表示起燃温度和完全转化温度。
[0096]
表1反应气的组成
[0097]
反应气的组成含量甲苯5000ppmo218%n281%co2333ppm其他4667ppm
[0098]
表2不同实施例、对比例与文献催化剂的甲苯催化燃烧的性能对比
[0099]
[0100][0101]
注:1.涂覆氧化石墨烯的堇青石载钯催化剂,源自文献(chinese journal of catalysis 39(2018)946
–
954)。2.二氧化铈(ceo2)载钯催化剂,反应条件为甲苯浓度1000pm,空速48000h-1
,源自文献applied catalysis b:environmental 220(2018)462
–
470.
[0102]
通过表1中各实施例催化剂与文献催化剂的对比,可以看出,本发明的催化剂起燃温度都降低了,完全转化温度也低于大多数文献值。其中,值得注意的是,文献pt/ceo2催化剂的测试条件与本发明不同,甲苯浓度更低、空速更高,这两个参数提高有利于甲苯催化燃烧,因此该催化剂的数据此处供参考。
[0103]
本发明所述的催化剂中,pd/cp催化剂活性最高,t
10
和t
90
都是最低。可以看出,在以木材为载体的催化剂中,钯与铂相比,性能更好。从木材种类来看,杨木和杉木相比,性能更好。
[0104]
从实施例1、2与对比例1、2的比较,可以看出,催化剂未经表面碳化处理,使得起燃温度与最终转化温度均升高,这说明表面碳化工艺对催化剂性能起到重要作用。本发明通过所述的tem和xrd研究发现,采用本发明所述的工艺条件配合创新的表面可控碳化手段,能够保持原有细胞阵列结构,且在其表面原位形成生长碳膜以及氧化石墨烯包覆的贵金属颗粒,该特殊的结构能够意外地改善催化活性。另外,从实施例5和对比例3可以看出,碳化
温度过低,也达不到应有的效果。当碳化温度过高(如500℃),容易引起木材骨架结构的坍塌,且导致细胞阵列结构整体碳化,容易导致催化性能大幅降低。因此在本发明所要求的温度下仅进行表面碳化,能够意外地维持细胞阵列结构,且可以意外地改善催化活性。
[0105]
从实施例1与对比例4、5的对照中可以看出,本发明的催化剂的制备工艺需要严格遵循“浸渍-还原-原位碳化”的顺序。如果改变顺序,都无法获得本发明保护的催化剂的结构,因而使得催化剂的性能降低。
[0106]
从实施例1与对比例6、7的对照中可以看出,原生木材粉末改变了其孔道结构,不利于催化剂的性能,这也说明了原生木材的细胞阵列孔道结构配合本发明所述的特殊的工艺,能够意外地协同改善催化剂的催化性能。另外,当采用虽具有纵向孔道,但横向致密的竹子为载体,也不利于获得高活性催化剂。这说明本发明采用的阔叶林原生木材,具有其结构独特性,和本发明所述的工艺具有意外的协同效果。
[0107]
另外,与二氧化铈相比,木材原生结构做载体也可以获得接近的金属分散度。我们测定了实施例1和实施例2的催化剂上钯的平均粒径分别为2.21和2.35nm。与pt/ceo2上的1.5-2.5nm粒径接近。本发明以大自然中的可再生资源
---
木材为载体,在粒径分布上获得与其他稀土金属氧化物类似的效果。
[0108]
实施例7
[0109]
pd/cp催化剂(同实施例1)在甲苯催化燃烧连续测试中的稳定性
[0110]
将实施例1的pd/cp催化剂,按照实施例5的反应气组成,在150℃进行长时间稳定性测试。结果显示,甲苯处理前浓度为5000ppm,经过催化处理后浓度降低为50ppm,转化率维持在99%,达到国家废气排放标准《大气污染物综合排放标准》gb16297-1996。经过168小时的连续运行,没有发现催化剂的活性降低。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1