一种非贵金属复合氧化物催化剂及其制备方法、应用
1.本技术涉及一种非贵金属复合氧化物催化剂,属于催化剂领域。
背景技术:
2.随着烟气脱硫脱硝设施的普遍应用,烟气中so2、nox的排放总量得到了有效控制。烟气中同时存在一定浓度的一氧化碳(co),近年来,烟气中co的排放问题日益受到重视。众所周知,co对人类和动物是一种有毒的气体,非常容易和血液中的血红蛋白结合削弱血红蛋白的输氧能力。co对大气环境有着重要的影响:co会与vocs和nox—起发生光化学反应,从而引起臭氧浓度的升髙;co是大气平流层中最重要活性物质oh自由基的控制气体,能控制oh与大气中几乎所有的痕量气体反应,从而进一步影响大气环境。
3.燃煤锅炉烟气中co的含量会随氧气过剩系数的变化而改变,火力电厂大型燃煤锅炉中co含量通常在100-1300mg/m3左右。而工业锅炉由于低的燃烧效率,烟气中co含量会更高。在钢铁行业中,焦炉烟气中co的浓度为2000mg/m3,烧结机尾气中co的含量更是高达10000mg/m3,如此高浓度的co排放至大气中,势必会对大气环境及生物体产生不利的影响。
4.目前脱硝最常用的方法是以nh3为还原剂的选择性催化还原方法(nh
3-scr),在脱硝反应器内,烟气中的no与喷入的nh3在催化剂表面反应生成n2和h2o。而脱除co的最高效常用的方法则是催化氧化,在催化剂的作用下,co可以被烟气中的o2氧化生成co2。如能实现在同一催化剂表面同时进行no的scr反应和co的氧化反应,则可利用现有的脱硝反应装置,在不额外增加设备的条件下,实现nox和co的一体化脱除。
5.中国专利cn104936679b给出了一种用于从烟道气或废气中同时除去一氧化碳和氮氧化物的方法和催化剂,其特征是在传统钒钨钛脱硝催化剂表面负载一层含有钯的贵金属催化剂,混合烟气通过催化剂时,co先被氧化,然后no可以被nh3还原。该催化剂使用了价格昂贵的贵金属,不适合于烟气量排放巨大的固定源烟气治理应用。
技术实现要素:
6.本技术提供了一种非贵金属复合氧化物催化剂,不同于现有技术中将两种不同功能的催化剂进行物理耦合实现催化不同的化学反应,而是在同一催化剂表面构筑不同的反应活性位点,实现复杂烟气中多种污染物的一步法同时脱除。该非贵金属复合氧化物催化剂可同时催化氨选择性还原氮氧化物反应和一氧化碳氧化反应,实现在同一催化床层上no和co的同时催化消除。
7.根据本技术的一个方面,提供了一种非贵金属复合氧化物催化剂。该非贵金属复合氧化物催化剂在同一催化剂表面构筑不同的反应活性位点,实现复杂烟气中多种污染物的一步法同时脱除。
8.一种非贵金属复合氧化物催化剂,包括第一组分、第二组分和第三组分;
9.所述第一组分选自cr2o3、v2o5、moo3、wo3、nb2o5中的至少一种;
10.所述第二组分选自mno2、co2o3、fe2o3、nio、cuo中的至少一种;
11.所述第三组分选自氧化铈、铈锆复合氧化物中的至少一种。
12.本技术中,当各组分选择两种或者两种以上时,对不同物质的比例不做严格限定。
13.铈-锆复合氧化物由于较小离子半径的zr
4+
取代了离子半径较大的ce
3+
,一方面可形成更多缺陷和晶格应力,另一方面可以补偿由ce
4+
向ce
3+
变化引起的体积膨胀,从而降低了氧离子扩散的活化能,有利于体相氧的迁移和扩散,提高了低温氧化还原性能。
14.可选地,以非贵金属复合氧化物催化剂的总质量为基准,所述第一组分的质量含量为1%~30%,所述第二组分的质量含量为1%~20%,所述第三组分的质量含量为50%~98%。
15.可选地,以非贵金属复合氧化物催化剂的总质量为基准,所述第一组分的质量含量为3%~10%,所述第二组分的质量含量为3%~10%,所述第三组分的质量含量为80%~94%。
16.可选地,所述第一组分的质量含量独立地选自1%、2%、2.7%、3%、5%、8.3%、8.5%、8.6%、8.7%、9%、9.3%、10%、15%、20%、30%中的任意值或任意两者之间的范围值。
17.可选地,所述第二组分的质量含量独立地选自1%、2%、3%、4%、4.3%、5%、5.4%、6%、6.3%、6.4%、6.8%、7%、8%、8.3%、9%、10%、15%、20%中的任意值或任意两者之间的范围值。
18.可选地,所述铈锆复合氧化物中,铈与锆的原子摩尔比为1:10~10:1。
19.优选地,铈与锆的原子摩尔比为1:1~4:1。
20.可选地,所述铈与锆的原子摩尔比独立地选自1:10、1:8、1:6、1:5、1:1、2:1、3:1、4:1、6:1、8:1、10:1中的任意值或任意两者之间的范围值。
21.根据本技术的另一个方面,提供了一种上述非贵金属复合氧化物催化剂的制备方法。
22.一种非贵金属复合氧化物催化剂的制备方法,包括如下步骤:
23.(1)获得第三组分;
24.(2)通过浸渍法将第一组分和第二组分浸渍在第三组分上。
25.可选地,步骤(1)为通过均相沉淀法制备氧化铈和/或铈锆复合氧化物粉末。
26.可选地,步骤(1)至少包括:向含有铈盐和/或锆盐的溶液中加入沉淀剂,沉淀,分离,水洗,干燥,焙烧,获得所述氧化铈和/或铈锆复合氧化物粉末。
27.可选地,所述铈盐选自硝酸铈、硝酸铈铵、氯化铈中的至少一种。
28.可选地,所述锆盐选自硝酸锆、硝酸氧锆、氧氯化锆中的至少一种。
29.具体地,制备含铈盐或铈盐加锆盐和尿素的水溶液,将此溶液加热沸腾一段时间后出现沉淀,过滤、洗涤、干燥后制得氧化铈或铈锆复合氧化物的前体。煅烧此前体可以得到氧化铈或铈锆复合氧化物。
30.可选地,步骤(2)至少包括:
31.(a)将第三组分的粉末溶于水中,得到悬浮液;
32.(b)将第一组分的前驱体和第二组分的前驱体加入上述悬浮液中,搅拌加热,得到糊状物;
33.(c)将所述糊状物干燥,煅烧,得到所述非贵金属复合氧化物催化剂。
34.可选地,所述第一组分的前驱体选自第一组分中含有的金属对应的可溶性盐。
35.可选地,所述第二组分的前驱体选自第二组分中含有的金属对应的可溶性盐。
36.可选地,所述第一组分的前驱体选自硝酸铬、偏钒酸铵、钼酸铵、偏钨酸铵、仲钨酸铵、草酸铌铵中的至少一种。
37.可选地,所述第二组分的前驱体选自硝酸锰、醋酸锰、硝酸钴、硝酸铁、硝酸镍、硝酸铜中的至少一种。
38.可选地,步骤(c)中,所述煅烧的条件为:温度450~600℃,时间2~4h。
39.具体地,第一组分和第二组分通过共浸渍的方式负载在第三组分上,首先制备氧化铈或铈锆复合氧化物粉末的含水悬浮液,搅拌状态下将第一组分和第二组分的前驱体盐加入上述悬浮液中,然后维持搅拌条件将所得混合物加热至大部分溶剂蒸发,得到一糊状物;干燥焙烧此糊状物后得到催化剂粉体材料。
40.作为一种具体的实施方式,非贵金属复合氧化物催化剂的制备方法,包括如下步骤:
41.1)采用均相沉淀法制备氧化铈或铈锆复合氧化物粉末;
42.2)制备氧化铈或铈锆复合氧化物粉末的含水悬浮液;
43.3)将第一组分和第二组分的前驱体加入上述悬浮液中;
44.4)将步骤3)所得混合物加热至大部分溶剂蒸发,得到一糊状物;
45.5)将步骤4)所得糊状物干燥和煅烧,得到催化剂粉末。
46.根据本技术的另一个方面,提供了一种上述非贵金属复合氧化物催化剂、所述的非贵金属复合氧化物催化剂的制备方法制备得到的非贵金属复合氧化物催化剂在同时催化氨选择性还原氮氧化物反应和一氧化碳氧化反应中的应用。
47.上述所述的非贵金属复合氧化物催化剂、上述所述的非贵金属复合氧化物催化剂的制备方法制备得到的非贵金属复合氧化物催化剂在同时催化氨选择性还原氮氧化物反应和一氧化碳氧化反应中的应用。
48.可选地,所述应用为在消除烟气排放的氮氧化物和一氧化碳中的应用。
49.可选地,所述烟气来源是固定源。
50.可选地,将含有氮氧化物、一氧化碳、氧气和氨气的混合气通入含有非贵金属复合氧化物催化剂的反应器,反应;
51.所述反应的条件为:体积空速为40000~100000h-1
,温度为150~500℃;
52.优选地,所述反应的条件为:体积空速为50000~100000h-1
,温度为150~500℃;
53.优选地,体积空速为40000~80000h-1
,温度为200~300℃。
54.作为一种具体的实施方式,提供了一种固定源烟气的净化方法,所述方法至少包括:将含有氮氧化物、一氧化碳和氨气的混合气在所述的非贵金属复合氧化物催化剂的存在下进行净化处理。
55.本技术提供的非贵金属复合氧化物催化剂用于固定源烟气净化系统中的氮氧化物选择性催化还原nh
3-scr过程和一氧化碳的催化氧化过程,在烟气中加入氨气后,在一定的温度区间,no和co可同时被消除,脱除效率可同时达到90%以上。
56.本技术能产生的有益效果包括:
57.1)本技术所提供的非贵金属复合氧化物催化剂,通过对第三组分氧化铈或铈锆复
合氧化物进行高价(hvd)和低价(lvd)金属氧化物掺杂改性,调变催化剂表面酸碱性,构建多种过渡金属元素间的氧化还原循环,实现在同一催化剂上no和co的同时催化消除。通过不同过渡金属元素的组合,构筑多个氧化还原循环催化剂体系,得到可在不同温度窗口起作用的no和co一体化消除催化剂。
58.2)本技术所提供的非贵金属复合氧化物催化剂,用于烟气治理,可以大大简化治理流程,降低污染物治理成本。采用常规分步脱除no及co的方法,需设置不同的反应单元,安装不同的催化剂,势必会增加场地面积、设备投资和运行成本。本技术提供的no和co一体化消除非贵金属复合氧化物催化剂可有效地规避上述问题,该催化剂即可以高效催化no和nh3的scr反应,同时也可以高效催化co的氧化反应,在一个反应器内一步完成no和co的高效脱除,使治理过程更为紧凑高效。
59.3)本技术所提供的非贵金属复合氧化物催化剂,不含贵金属,成本较低,可适用于烟气量巨大的固定源烟气污染物治理。
附图说明
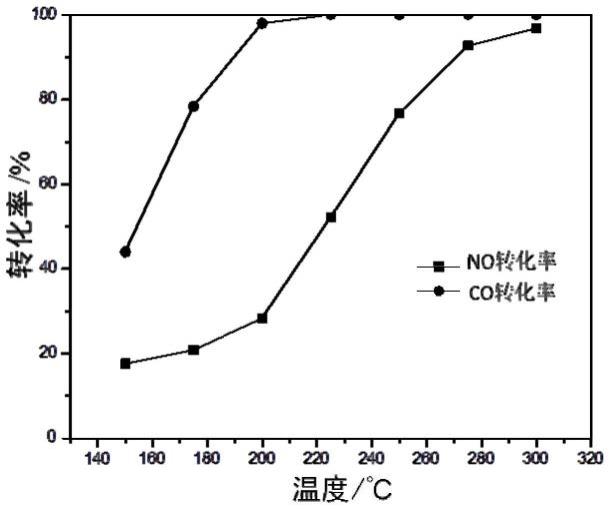
60.图1为实施例1制备的6.4%cuo-8.5%wo3/ceo2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
61.图2为实施例2制备的6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
62.图3为实施例3制备的5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
63.图4为实施例4制备的8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
64.图5为实施例5制备的4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
65.图6为实施例6制备的4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
66.图7为实施例7制备的8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
67.图8为实施例8制备的6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
68.图9为实施例9制备的10%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
69.图10为实施例10制备的5%cuo-10%wo3/ce
0.8
zr
0.2
o催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
70.图11为实施例11制备的10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
71.图12为对比例1制备的10%cuo/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
72.图13为对比例2制备的10%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口
气体温度变化曲线。
73.图14为对比例3制备的10%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
具体实施方式
74.下面结合实施例详述本技术,但本技术并不局限于这些实施例。
75.如无特别说明,本技术的实施例中的原料均通过商业途径购买。如无特别说明,测试方法均采用常规方法,仪器设置均采用均厂家推荐的设置。
76.本技术的实施例中脱硝效率、脱co效率计算如下:
77.其中nox
out
为氮氧化物出口浓度,nox
in
为氮氧化物入口浓度。
78.其中co
out
为一氧化碳出口浓度,co
in
为氮氧化物入口浓度。
79.实施例中各组分百分比均为质量比,以非贵金属复合氧化物催化剂的总质量为基准。
80.实施例1 6.4%cuo-8.5%wo3/ceo2催化剂的制备及活性评价
81.取86.8g硝酸铈ce(no3)3·
6h2o和180g尿素置于烧杯中,加去离子水溶解制成2l混合溶液。在搅拌条件(500rpm)下加热此混合溶液至沸腾。溶液沸腾一定时间后变混浊,出现沉淀。继续沸腾状态下搅拌2小时后,将得到的混合物进行抽滤使沉淀与母液分离;用去离子水洗涤沉淀两次,最后滤饼抽干后转入烘箱内,在100℃下烘干15小时,然后放入马弗炉中,500℃焙烧2小时得到ceo2粉体。
82.采用浸渍法制备6.4%cuo-8.5%wo3/ceo2催化剂。首先在搅拌条件下将15g ceo2粉末混悬在50ml去离子水中,然后依次加入3.39gcu(no3)2·
3h2o和1.76g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为6.4%,wo3含量为8.5%(重量比)的6.4%cuo-8.5%wo3/ceo2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
83.将1.15g样品装入石英管中,反应气氛为:500ppm no+500ppm nh3+1000ppmco+10%o2+10%h2o+n2,调节原料气流量使得反应的空速为50000h-1
。于不同温度下分别检测进出口的no、no2和co浓度,从而计算出总的no
x
转化率和co转化率,得到催化剂的脱硝效率和脱co效率。图1为实施例1制备的6.4%cuo-8.5%wo3/ceo2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
84.实施例2 6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5
o2催化剂制备及活性评价
85.取54.8g硝酸铈铵(nh4)2ce(no3)6、100ml 1m的硝酸锆溶液和180g尿素置于烧杯中,加去离子水溶解制成2l混合溶液。在搅拌条件(500rpm)下加热此混合溶液至沸腾。溶液沸腾一定时间后变混浊,出现沉淀。继续沸腾状态下搅拌2小时后,将得到的混合物进行抽滤使沉淀与母液分离;用去离子水洗涤沉淀两次,最后滤饼用异丙醇淋洗抽干后转入真空
烘箱内,在60℃下烘干15小时,然后放入马弗炉中,500℃焙烧2小时得到ce/zr摩尔比为1:1的ce
0.5
zr
0.5
o2粉体。
86.采用浸渍法制备6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5
o2催化剂。首先在搅拌条件下将20g ce
0.5
zr
0.5
o2粉末混悬在50ml去离子水中,然后依次加入4.52gcu(no3)2·
3h2o和11.55gcr(no3)3·
9h2o。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为6.3%,cr2o3含量为9.3%(重量比)的6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
87.6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5
o2催化剂的活性评价条件同实施例1。图2为实施例2制备的6.3%cuo-9.3%cr2o3/ce
0.5
zr
0.5o22
催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
88.实施例3 5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂制备及活性评价
89.称取393gzr(no3)4·
3h2o置于烧杯中加入一定量的去离子水加热溶解,定容至1l得到1m的硝酸锆溶液。取87.68g硝酸铈铵(nh4)2ce(no3)6、40ml 1m的硝酸锆溶液和180g尿素置于烧杯中,加去离子水溶解制成2l混合溶液。在搅拌条件(500rpm)下加热此混合溶液至沸腾。溶液沸腾一定时间后变混浊,出现沉淀。继续沸腾状态下搅拌2小时后,将得到的混合物进行抽滤使沉淀与母液分离;用去离子水洗涤沉淀两次,最后滤饼用异丙醇淋洗抽干后转入真空烘箱内,在60℃下烘干15小时,然后放入马弗炉中,500℃焙烧2小时得到ce/zr摩尔比为4:1的ce
0.8
zr
0.2
o2粉体。
90.采用浸渍法制备5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入4.11g50wt%硝酸锰水溶液和1.17g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到mno2含量为5.4%,wo3含量为8.6%(重量比)的5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
91.5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图3为实施例3制备的5.4%mno
2-8.6%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
92.实施例4 8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
93.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入3.88gni(no3)2·
3h2o和1.17g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到nio含量为8.3%,wo3含量为8.3%(重量比)的8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
94.8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图4为实施例4制备的8.3%nio-8.3%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
95.实施例5 4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂制备及活性评价
96.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入1.51gcu(no3)2·
3h2o和2.28g草酸铌铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为4.3%,nb2o5含量为8.7%(重量比)的4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
97.4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图5为实施例5制备的4.3%cuo-8.7%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
98.实施例6 4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂制备及活性评价
99.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入1.51gcu(no3)2·
3h2o和1.23g钼酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为4.3%,moo3含量为8.7%(重量比)的4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
100.4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图6为实施例6制备的4.3%cuo-8.7%moo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
101.实施例7 8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂制备及活性评价
102.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入3.89gco(no3)2·
6h2o和1.23g钼酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到coo含量为8.3%,moo3含量为8.3%(重量比)的8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
103.8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图7为实施例7制备的8.3%coo-8.3%moo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
104.实施例8 6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
105.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂。首先称取4.63g偏钒酸铵和7.13g草酸加入去离子水溶解,定容至100ml得到偏钒酸铵溶液。在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在40ml去离子水中,然后依次加入2.26gcu(no3)2·
3h2o和8.3ml上述配制的偏钒酸铵溶液。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为6.8%,v2o5含量为2.7%(重量比)的6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
106.6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例1。图8为实施例
8制备的6.8%cuo-2.7%v2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
107.实施例9 10%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
108.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备10%cuo-10%wo3/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入3.79gcu(no3)2·
3h2o和1.34g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为10%,wo3含量为10%(重量比)的10%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
109.10%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件基本同实施例1,只是催化剂的装填量为0.9g,并调节气体流量使得反应空速为80000hr-1
。图9为实施例9制备的10%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
110.实施例10 5%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
111.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备5%cuo-10%wo3/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入1.79gcu(no3)2·
3h2o和1.29g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为5%,wo3含量为10%(重量比)的5%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
112.5%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例9,图10为实施例10制备的5%cuo-10%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
113.实施例11 10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2催化剂制备及活性评价
114.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后依次加入3.80gcu(no3)2·
3h2o和2.85g草酸铌铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为10%,nb2o5含量为10%(重量比)的10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
115.10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例9,图11为实施例11制备的10%cuo-10%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
116.对比例1 10%cuo/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
117.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备10%cuo/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后加入3.37gcu(no3)2·
3h2o。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到cuo含量为10%的10%cuo/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
118.10%cuo/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例9,图12为对比例1制备的
10%cuo/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
119.对比例2 10%wo3/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
120.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备10%wo3/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后加入1.18g偏钨酸铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到wo3含量为10%(重量比)的10%wo3/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
121.10%wo3/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例9,图13为对比例2制备的10%wo3/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
122.对比例3 10%nb2o5/ce
0.8
zr
0.2
o2催化剂的制备及活性评价
123.ce
0.8
zr
0.2
o2粉体的制备同实施例3。采用浸渍法制备10%nb2o5/ce
0.8
zr
0.2
o2。首先在搅拌条件下将10g ce
0.8
zr
0.2
o2粉末混悬在50ml去离子水中,然后加入2.51g草酸铌铵。在磁力搅拌加热器上持续搅拌加热,使得大部分水被蒸发得到一糊状物。将糊状物转入烘箱,120℃干燥12小时。干燥样品在500℃下煅烧2小时,得到wo3含量为10%(重量比)的10%nb2o5/ce
0.8
zr
0.2
o2催化剂。将粉末压片、破碎筛分后得到40~60目颗粒供活性评价用。
124.10%nb2o5/ce
0.8
zr
0.2
o2催化剂的活性评价条件同实施例9,图14为对比例3制备的10%nb2o5/ce
0.8
zr
0.2
o2催化剂上no
x
转化率和co转化率随入口气体温度变化曲线。
125.由上述非贵金属复合氧化物催化剂的评价结果曲线可以看出,通过第一组分和第二组分的不同组合,本技术的三组分非贵金属复合氧化物催化剂,在一定的温度区间,no和co的转化率均可达到90%以上,显示了良好的同时脱除no和co的催化活性,可适用于不同工况下的烟气进行no和co的脱除治理。
126.以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1