壳聚糖/纤维素复合微球固载铜在催化α,β-不饱和羰基化合物的硅加成反应中的应用
壳聚糖/纤维素复合微球固载铜在催化
α
,
β
‑
不饱和羰基化合物的硅加成反应中的应用
技术领域
1.本发明涉及一种催化材料的制备及在有机硅化合物中的应用,具体涉及一种壳聚糖/纤维素复合微球固载铜催化材料(chitosan/cellulose
‑
cu
2+
=cc@cu)的制备方法及在α,β
‑
不饱和羰基化合物的硅加成反应中的应用。
背景技术:
2.有机硅化合物是一类重要的中间体,广泛应用于天然产物与药物分子的合成中,因为c
‑
si键比较稳定可以作为合成过程中的保护基团,不会发生分解或副反应。相较于传统所报道的使用强碱的制备方法,在催化剂作用下,对不饱和羰基化合物直接硅加成的策略更为直接和有效,近年来获得广泛关注。文献中催化硅加成的反应所使用的催化剂为pd(chem.commun.,2016,52,5609
‑‑
5612),rh(angew.chem.int.ed.2006,45,5675
–
5677)等,但是这些报道中多使用贵金属,成本高不适用于实际生产。因此铜作为一种来源广泛、廉价易得的过渡金属在催化硅加成的反应中就体现出了其独特的优势,2010年,文献(j.am.chem.soc.2010,132,2898
–
2900)报道使用cucl为催化剂,添加强碱naot
‑
bu,在
‑
78℃低温条件下,无需质子源存在的情况下,实现了(二甲基苯硅烷基)联硼酸频那醇酯me2phsi
‑
bpin对α,β
‑
不饱和羰基化合物的β
‑
硅加成反应,该方法使用了金属一价铜盐,且无需质子源,但伴随的是强碱的使用、低温条件以及需要使用价格昂贵的nhc配体,后处理复杂,对环境不友好。2015年,文献(j.am.chem.soc.2015,137,15422
‑
15425)报道使用cu(acac)2为催化剂与特制手性联吡啶配体作用,以h2o为溶剂,室温条件下,催化制备有机硅化合物,方法简单但是该方法配体制备方法复杂,未实现商业化,限制反应成本,不利于实际生产,同时存在催化剂无法回收利用的问题;2018年,文献(j.chin.chem.soc.2018,65,81
–
86)中使用cu2(oh)2co3为催化剂同时添加2,2
‑
联吡啶配体和表面活性剂triton x
‑
100相互作用,以纯水为溶剂,室温条件反应,但也存在着反应成本限制、催化剂无法回收利用等问题。以上方法虽然反应的活性有所提高,但同时也存在着反应条件限制、成本高昂、环境污染以及催化剂无法回收利用等问题,这些都极大的限制了此类方法在实际生产中的应用。因此,发展一种简便易操作、条件温和、成本低廉,以直接硅加成策略制备有机硅化合物的绿色环保的新方法是十分迫切需要的。
3.有机硅化合物向β
‑
羟基化合物的转化是有机硅化合物的一类重要应用,因为β
‑
羟基结构广泛存在于天然产物结构之中,因此若能采用“一锅法”的策略,首先实现底物的硅加成,之后不需分离连续转化为β
‑
羟基化合物,将简化天然产物的合成步骤,具有十分重要的应用价值。此外,有机硅化物自身在有机合成和材料化学中也有广泛的实际用途,例如聚合反应的单体、抗癌药物的前驱体、偶联剂等。
技术实现要素:
4.本发明提供一种壳聚糖/纤维素复合微球固载铜金属催化材料(cc@cu)的制备方
法及将其应用于制备有机硅化合物及β
‑
羟基化合物的方法,旨在至少一定程度上克服现有技术中存在的如下不足:以贵金属为合成有机硅化合物的催化剂或昂贵的硅试剂为合成原料时,成本高,无法工业化;以一价铜和氮卡宾配体为催化剂时,操作过程复杂,需要强碱(叔丁醇钾等)、低温(
‑
78℃),严格无水等苛刻条件,同样造成生产成本较高;现有的β
‑
羟基化合物制备时若以有机硅化合物为起点,则意味着合成有机硅化合物后需要将其从反应产物中进行分离提纯处理,没有连续化生产,故工艺路线复杂,生产效率较低。本发明以壳聚糖/纤维素复合微球固载铜催化材料制备有机硅化合物,利用壳聚糖/纤维素复合微球固载铜催化材料的独特的相容性和空间结构,具有更大的比表面积,催化活性更高,能在纯水中实现催化反应,同时能多次回收利用,符合绿色化学的理念,非常适合工业化应用。
5.本发明解决上述技术问题的技术方案如下:
6.壳聚糖/纤维素复合微球固载铜在催化α,β
‑
不饱和羰基化合物的硅加成反应中的应用,包括以下步骤:
7.1)将α,β
‑
不饱和羰基化合物i、联硼酸频那醇二甲基硅试剂和壳聚糖/纤维素复合微球固载铜催化材料cc@cu按照摩尔比1:1.2:0.01的比例加入到水中,cc@cu与水的用量之比为0.002mmol:1ml,在室温下搅拌12h,发生硅加成反应:
[0008][0009]
其中,r1为苯酮基、对甲氧基苯酮基、对甲基苯酮基、对卤代苯酮基、乙酰基、甲醛基、甲酯基、乙酯基或氰基;r2为苯基、对甲基苯基、对卤代苯基、邻卤代苯基、对甲氧基苯基、甲基、叔丁基、乙酰基、对卤代甲基苯基;
[0010]
2)将所述cc@cu过滤,萃取后将滤液溶剂旋干,用薄层层析方法分离产物,得到有机硅化合物ii;
[0011]
所述cc@cu是壳聚糖和纤维素的混合溶液混合后在碱性溶液中形成微球,然后加入致孔剂、交联剂交联形成复合微球,再浸滞在cu
2+
的水溶液中得到的催化剂,铜在所述cc@cu中的相对含量为1.75
×
10
‑3mol/g,含有醛或酮的溶液中c=o和壳聚糖的单元体的摩尔比为8~12:1。
[0012]
前述的应用中,所述cc@cu的制备方法包括以下步骤:
[0013]
1)将纤维素颗粒壳聚糖溶液中至搅拌均匀,纤维素与壳聚糖的质量比为400mg:1.5g,将所制得的混合溶液用注射器向氢氧化钠溶液中缓慢滴入形成透明状微球;
[0014]
2)通过过滤回收微珠,用蒸馏水和乙醇充分洗涤,加入含有乙醇和含有醛或酮的溶液中于50℃搅拌12小时,进行交联,含有醛或酮的溶液中c=o和壳聚糖的单元体的摩尔比为8~12:1;
[0015]
3)滤出交联后黄棕色复合微珠,用水和乙醇冲洗,在室温下进行干燥;
[0016]
4)将干燥后的微球在50℃的水中浸泡悬浮1小时;将cu
2+
的水溶液加入该悬浮液中缓慢搅拌12h,进行吸附铜离子;
[0017]
5)通过过滤分离负载cu
2+
的微球,用水和乙醇冲洗以除去游离的铜离子和阴离子,
最后,将cc@cu在50℃下烘箱干燥12小时,得到cc@cu催化材料。
[0018]
前述的应用中,所述α,β
‑
不饱和羰基化合物i为查尔酮。
[0019]
前述的应用中,步骤2)中:将所述cc@cu过滤后,用水和乙醇充分洗涤3次,而后进行干燥进行重复使用。
[0020]
前述的应用中,含有醛或酮的溶液中c=o和壳聚糖的单元体的摩尔比为8:1。
[0021]
前述的应用中,所述cc@cu循环连续使用6次后,第7次应用于所述查尔酮的硼加成反应,产物的收率为80%。
[0022]
推测可能的反应机理如下:
[0023]
首先,联硼酸频哪醇二甲基硅试剂[phme2si
‑
b(pin)]在cc@cu催化材料的中活性铜的催化下发生si
‑
b键的断裂(催化材料中的壳聚糖上含有大量的氨基,为反应提供了一种碱性环境),与二价铜反应形成铜硅烷基络合物以及副产物bpin
‑
oh,中间体在羰基的导向作用下,与α,β
‑
不饱和羰基化合物发生共轭加成,而后经过六元环过渡重排,在水提供质子源的作用下发生质子化过程生成目标产物,并实现催化材料的再生,在反应中,水即为质子源也为溶剂。
[0024]
与传统方法相比,本发明具有以下优点:
[0025]
1.壳聚糖/纤维素复合微球具有良好的生物相容性,绿色环保,可用于参加纯水反应,固载金属铜效果好,同时具有较长的的使用寿命,壳聚糖/纤维素复合微球固载铜催化材料在反应完成后可方便地借助固液分离方法与反应体系中其他组分分离,经过简单的再生就可以重复使用,因此可大大降低生产成本,同时也可以明显减少各种环境污染问题。
[0026]
2.该方法仅需要使用较低的催化剂用量,即可实现反应物较高的转化率;
[0027]
3.该方法反应条件温和,以纯水为溶剂,在室温下进行反应,简便易操作;
[0028]
4.该方法应用性广,可用于各种不同类型的α,β
‑
不饱和羰基化合物的硅加成,成功制备出相应的有机硅化合物及β
‑
羟基化合物。
[0029]
5.该方法中可采用“一锅法”的策略,起始原料经连续的硅加成反应、氧化反应直接制备出含有羰基的β
‑
羟基化合物。
[0030]
6.仅以壳聚糖负载二价铜为催化剂时,在纯水中进行α,β
‑
不饱和受体硼加成反应时,以查尔酮为模板底物、壳聚糖负载氢氧化铜为催化剂、2,2
‑
联吡啶为配体,与b2(pin)2在纯水中反应,其中壳聚糖固载氢氧化铜(cs@cu(oh)2)和壳聚糖固载氧化铜(cs@cuo)依文献(carbohydrate polymers2015,134,190
‑
204)制备,壳聚糖固载氰化铜(cs@cucn),壳聚糖固载硫酸铜(cs@cuso4),壳聚糖固载氯化铜(cs@cucl2),壳聚糖固载氟化铜(cs@cuf2)和壳聚糖固载溴化铜(cs@cubr2)依文献(greenchem.2014,16,3007
‑
3012)制备。壳聚糖对cu
2+
的吸附以氨基配合[(2)式]为主,主要吸附反应包括:
[0031]
氨基质子化:
[0032]
配合:
[0033]
氢键吸附:
[0034]
静电吸引:
[0035]
ph较低时,参与质子化反应形成的
‑
nh
3+
数较多,而用于配合吸附cu
2+
的
‑
nh2较少。通过icp检测反应后回收的催化剂(cs@cu(oh)2)发现有1.6%的铜脱落,考虑到氢氧化铜不
溶于水,推测可能是(pin)boh副产物的生成导致体系ph值改变,进一步对二价铜盐的种类进行筛选,发现当使用水溶性较好的铜盐(如硫酸铜、氯化铜)时,铜的脱落更为严重,因此,转而采用添加配体的策略来调整反应体系的ph值,最终发现含酰胺键的吡啶类配体可以很好地抑制铜的脱落,起到稳定催化剂的作用。
[0036]
本技术经过壳聚糖和纤维素复合交联后,提高催化剂载体的致密度,增加微孔结构,壳聚糖上的伯胺(r’nh2)与醛酮(r2c=o)发生schiff碱反应,生成含碳氮双键的亚胺(r2c=nr’),醛酮在壳聚糖分子内部和分子间不同氨基位形成桥连接,schiff碱反应虽然减少了壳聚糖表面的氨基,但由于壳聚糖表面富含羟基,schiff碱反应形成的c=n双键中的n原子与邻近oh中的o原子极易与cu
2+
发生络合,形成共轭平面(xie x j,qin y.sens actuators b,2011,156(1):213),交联还提高了壳聚糖的耐酸能力,通过化学吸附和物理吸附,对铜离子的络合作用更强。
[0037]
7.以壳聚糖与醛或酮交联时,由于会发生缩醛化反应,醛或酮的
‑
c=o相对于壳聚糖单元体的
‑
nh2大幅过量时,才能生成足够的亚胺基,用于与铜离子形成稳定的络合物。但过于过量时又因缩醛反应使与c=n双键中的n原子邻近oh中的o原子减少,降低反应物产率,故制备cc@cu是壳聚糖和纤维素的交联复合微球时,交联剂中醛或酮的溶液中c=o和壳聚糖的单元体的摩尔比为8~12:1较适宜。
附图说明
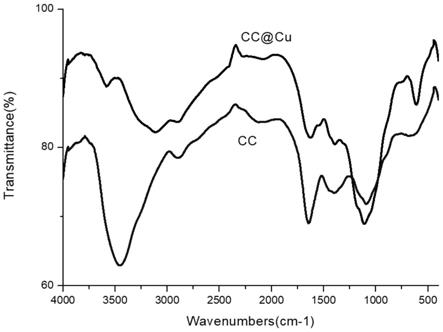
[0038]
图1为负载二价铜离子的交联壳聚糖/纤维素复合微球cc@cu和交联壳聚糖/纤维素的微球的红外光谱图;
[0039]
图2为壳聚糖与酮或醛交联反应的机理图。
具体实施方式
[0040]
以下结合具体实施例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0041]
实施例1:
[0042]
本发明实施例提供的cc@cu催化材料的活性成分为铜,载体为壳聚糖/纤维素复合微球;同时,活性成分铜在cc@cu催化材料中的相对含量为1.75mmol/g。
[0043]
其中,载体为壳聚糖/纤维素复合微球,壳聚糖/纤维素复合微球为纤维素混入壳聚糖的酸溶液后加入碱性溶液中悬浮形成微球,再加入致孔剂、交联剂交联形成的复合微球。
[0044]
实施例2:
[0045]
本发明实施例还提供一种cc@cu催化材料的制备方法,包括三大步骤:
[0046]
1)壳聚糖/纤维素微球的制备:将纤维素颗粒(400mg)加入到100ml的壳聚糖溶液中(100ml水,1.5g壳聚糖,3.0ml醋酸)至搅拌均匀,得粘稠液。将所制得的混合溶液用注射器向100ml氢氧化钠溶液中缓慢滴入(由15g氢氧化钠,蒸馏水100ml配成)形成透明状微球。壳聚糖分子中的羟基和氨基有良好的反应活性,可以方便地进行接枝和改性,壳聚糖可通过配合、离子交换和静电作用吸附金属离子,但壳聚糖微球的强度和耐酸性差,壳聚糖由于质子化氨基的亲水性溶于酸,壳聚糖的分子式为(c6h
11
no4)n,单元体的分子量为:161.2,
1.5g壳聚糖含0.009moml单元体。纤维素抗拉强度高,与壳聚糖分子结构相似,具有相容性,通过纤维素与壳聚糖共混可提高强度,同时改善吸附剂孔结构和表面特性,有利于提高吸附性能。
[0047]
2)交联壳聚糖/纤维素微球的制备:通过过滤回收微珠,用蒸馏水和乙醇充分洗涤,在含有乙醇(100ml)和丁二醛(壳聚糖单元体:丁二醛=1mol:4mol)的交联溶液中于50℃搅拌12小时,滤出交联后的黄棕色复合微珠,用水和乙醇洗涤,在室温下干燥。本步骤中通过乙醇致孔,丁二醛交联制备出多孔交联壳聚糖/纤维素复合微球,交联后氨基下降,可提高壳聚糖在酸性介质中的化学稳定性。
[0048]
3)负载二价铜离子的交联壳聚糖/纤维素微球(cc@cu)的制备:将交联干燥后的微球(1.0g)在50℃的水(20ml)中悬浮浸泡1小时。将10ml硫酸铜溶液(由100mg五水硫酸铜配置,约0.0004moml)加入该悬浮液中并搅拌12h,通过过滤分离负载cu
2+
的微球,用水和乙醇冲洗以除去游离的铜离子和硫酸根离子。最后,将壳聚糖/纤维素
‑
cu
2+
(cc@cu)催化材料在50℃下烘箱干燥12小时,得到上述的cc@cu催化材料。
[0049]
图1为壳聚糖/纤维素复合微球(cc)和cc@cu催化材料的红外光谱图,位于上方的线代表壳聚糖/纤维素复合微球(cc)负载cu之前的红外光谱,下方的线代表壳聚糖/纤维素复合微球(cc)负载cu之后的红外光谱。
[0050]
壳聚糖/纤维素复合微球(cc)通过壳聚糖的
‑
nh2基与丁二醛的酮基缩合形成亚胺基(c=n),如图2中的i过程和iv过程。在1636cm
‑1观察到载体亚胺基团的拉伸,在3422.15cm
‑1观察到载体n
‑
h基团的拉伸。在催化剂cc@cu的表征曲线上,亚胺基团带在1621cm
‑1处的振动有所减弱,这可以归因于铜离子与配体亚胺基的配位。n
‑
h基团在3422.15cm
‑1处的振动消失,这归因于铜离子与n
‑
h基团的络合,所有n
‑
h基团都参与了与铜离子的络合,铜离子与配体亚胺基配位后,不易因ph值改变导致铜脱落。图1中(cc)和cc@cu在1110cm
‑1处均可观察到一个较强的新特征峰,它来自于c
‑
o
‑
c
‑
o
‑
c基团的伸展振动,这表明丁二醛中的羰基亦与壳聚糖葡萄糖胺环上的羟基发生了缩醛化反应,如图2中的ii过程,从理论上说,羰基可以与羟基进行缩醛化反应。将一摩尔的醛和一摩尔的醇混合在一起会形成可逆的反应平衡,其产物为半缩醛。半缩醛是由醇对羰基的亲核加成而生成的,其结构特征是一个
‑
oh基团和一个
‑
or连接在相同的碳原子上。半缩醛通常很不稳定而无法分离出来,它可以在少量气态盐酸的催化作用下进行第二次反应,与另一摩尔的醇反应,生成一摩尔缩醛。缩醛的特征是两个
‑
or基团连接在相同的ch基团上。缩醛化反应最可能发生在羰基与空间位阻相对较弱的壳聚糖c6羟基之间,缩醛的特征峰应该出现在交联壳聚糖纤维红外光谱图中的1105~116 0cm
‑1区间,因此1110cm
‑1特征峰的存在说明交联反应不仅发生在丁二醛和伯胺之间,还发生在丁二醛与羟基之间。但是此处峰强比1621~1636cm
‑1处(schiff碱特征峰)同样强,表明在交联反应中schff碱反应和缩醛化反应占主导地位。丁二醛与壳聚糖分子环上羟基反应的结构示意图见图2中的ii,从图中可见,一个丁二醛分子可以与四个羟基反应形成交联结构。(cc)和cc@cu中我们没有观察到任何可归因于丁二醛碳基团的谱带拉伸(在1750cm
‑1左右),证实了丁二醛的所有酮基都参与了席夫碱反应和缩醛化反应。交联反应中无丁二醛只有一端的羰基与壳聚糖分子环上的氨基进行了反应,而另一端的羰基未有反应的情况,从而形成了n=c
‑
o=o结构(见图2中的iv过程)。在3422.15cm
‑1观察到载体n
‑
h基团的拉伸也归因于丁二醛的用量不足,丁二醛未将全部的
‑
nh2转换为
‑
c=n。
[0051]
实施例3~5
[0052]
采用实施例2的方法制备cc@cu催化材料,不同在于,实施例3的第2)步中壳聚糖单元体:丁二醛=1mol:1mol,此时可能由于交联不完全,微球呈透明状,脆弱易变性,即排除这一比例;实施例4的第2)步中壳聚糖单元体:丁二醛=1mol:2mol,可能交联不完全,微球呈透明状,易变性;实施例5的第2)步中壳聚糖单元体:丁二醛=1mol:6mol,微球呈偏褐色实心状,形态稳定。结合图2和实施例2对催化材料的红外光谱分析,当丁二醛的
‑
c=o与壳聚糖单元体的
‑
nh2为2:1或4:1时交联不完全,可归因于丁二醛的所有酮基都参与了席夫碱反应和缩醛化反应,席夫碱反应和缩醛化反应都占主导地位,即使丁二醛的
‑
c=o相对于壳聚糖单元体的
‑
nh2过量,也不能保证
‑
nh2全部都发生席夫碱反应;丁二醛的
‑
c=o有相当部分被缩醛反应消耗掉了。
[0053]
实施例6:
[0054]
本发明实施例还提供一种将cc@cu催化材料应用于α,β
‑
不饱和羰基化合物和联硼酸频那醇二甲基硅试剂之间的硅加成反应,具体步骤如下:将α,β
‑
不饱和羰基化合物i、联硼酸频那醇二甲基硅试剂和cc@cu催化材料(实施例2制备)按照摩尔比1:1.2:0.01的比例加入到1.0ml水的混合溶剂中,cc@cu与水的用量之比为0.002mmol:1ml,在室温下搅拌12h;将cc@cu催化材料过滤后,萃取后旋干溶剂,通过薄层层析分离后,得到有机硅化合物ii。同时,cc@cu催化材料首次应用于α,β
‑
不饱和羰基化合物与联硼酸频那醇二甲基硅试剂之间的硅加成反应中。该硅加成反应如下:
[0055][0056]
其中,其中r1为苯酮基、对甲氧基苯酮基、对甲基苯酮基、对卤代苯酮基、乙酰基、甲醛基、甲酯基、乙酯基或氰基;r2为苯基、对甲基苯基、对卤代苯基、邻卤代苯基、对甲氧基苯基、甲基、叔丁基、乙酰基、对卤代甲基苯基,例如上述α,β
‑
不饱和羰基化合物为查尔酮,反应后将cc@cu催化材料过滤,用水和乙醇充分洗涤多次,而后进行干燥,便能进行重复使用。
[0057]
应用例1:
[0058]
将上述实施例2提供的cc@cu催化材料应用于查尔酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,查尔酮为0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
1(58.6mg),收率为85%。
[0059]
[0060]
目标产物的核磁氢谱和碳谱如下所示:
[0061]1h nmr(400mhz,chloroform
‑
d);δ=7.76
–
7.73(m,2h),7.48
–
7.44(m,1h),7.42
–
7.39(m,2h),7.36
–
7.28(m,5h),3.49(dd,j=17.1,10.2hz,1h),3.19
–
3.05(m,2h),0.26(s,3h),0.20(s,3h).
[0062]
13
c nmr(100mhz,chloroform
‑
d);δ=199.1,142.4,137.1,136.9,134.2,132.8,129.4,128.5,128.1,128.0,127.8,127.7,124.8,39.0,31.1,
‑
3.8,
‑
5.1.
[0063]
应用例1表明,在本发明实施例2提供的cc@cu催化材料的催化条件下,查尔酮的转化率很高,其硅加成产物的收率达到了85%。
[0064]
将实施例5制得的催化材料按以上反应步骤应用于α,β
‑
不饱和羰基化合物(查尔酮)和联硼酸频那醇二甲基硅试剂之间的硅加成反应,产率为68%。
[0065]
催化剂中更多的
‑
nh2并不能提高催化效率,随着丁二醛的用量增加,壳聚糖中更多的
‑
nh2发生席夫碱反应,壳聚糖交联的更完全,使铜离子在硅加成反应中与催化剂结合的更牢固,不因中间产物引起ph值降低而导致铜离子脱落,降低反应转化率。但当丁二醛的用量再进一步加大时,产率降低,可能归因于缩醛反应使壳聚糖葡萄糖胺环上的羟基减少,因此没有足够的邻近oh中的o原子配合c=n双键中的n原子与cu
2+
发生络合。
[0066]
应用例2:
[0067]
将上述实施例2提供的cc@cu催化材料应用于(1
‑
苯基
‑3‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(1
‑
苯基
‑3‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮为0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
2(50.2mg),收率为70%。
[0068][0069]
目标产物的核磁氢谱和碳谱如下所示:
[0070]1h nmr(400mhz,chloroform
‑
d);δ=7.79(d,j=7.6hz,2h),7.52
–
7.46(m,3h),7.40
–
7.33(m,5h),7.00(d,j=7.8hz,2h),6.89(d,j=7.7hz,2h),3.49(dd,j=17.0,10.4hz,1h),3.21
–
3.04(m,2h),2.27(s,3h),0.30(s,3h),0.23(s,3h).
[0071]
13
c nmr(100mhz,chloroform
‑
d);δ=199.2,139.1,137.1,137.1,134.2,134.1,132.8,129.3,128.9,128.5,128.0,127.8,127.6,77.3,39.0,30.6,21.0,
‑
3.7,
‑
5.2.
[0072]
应用例2表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(1
‑
苯基
‑3‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了70%。
[0073]
应用例3:
[0074]
将上述实施例2提供的cc@cu催化材料应用于(e)
‑3‑
(4
‑
溴苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(e)
‑3‑
(4
‑
溴苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮为0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
3(67.7mg),收率为80%。
[0075][0076]
目标产物的核磁氢谱和碳谱如下所示:
[0077]1h nmr(400mhz,chloroform
‑
d);δ=7.79
–
7.77(m,2h),7.54
–
7.49(m,1h),7.44
–
7.33(m,7h),7.28
–
7.26(m,2h),3.46(dd,j=17.2,10.6hz,1h),3.22
–
3.03(m,2h),0.30(s,3h),0.25(s,3h).
[0078]
13
c nmr(100mhz,chloroform
‑
d);δ=198.7,141.6,136.9,136.3,134.2,133.0,131.1,129.5,129.3,128.5,127.9,127.9,118.4,38.7,30.8,
‑
4.0,
‑
5.2.
[0079]
应用例3表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(e)
‑3‑
(4
‑
溴苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了80%。
[0080]
应用例4:
[0081]
将上述实施例2提供的cc@cu催化材料应用于(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮为0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
4(56.8mg),收率为75%。
[0082][0083]
目标产物的核磁氢谱和碳谱如下所示:
[0084]1h nmr(400mhz,chloroform
‑
d);δ=7.80
–
7.78(m,2h),7.54
–
7.50(m,1h),7.45
–
7.34(m,7h),7.15
–
7.13(m,2h),6.90
–
6.88(m,2h),3.48(dd,j=17.2,10.6hz,1h),3.23
–
3.08(m,2h),0.30(s,3h),0.26(s,3h).
[0085]
13
c nmr(100mhz,chloroform
‑
d);δ=198.8,141.0,136.9,136.3,134.2,133.0,130.4,129.5,128.9,128.6,128.2,127.94,127.90,38.7,30.7,
‑
4.0,
‑
5.2.
[0086]
应用例4表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了75%。
[0087]
应用例5:
[0088]
将上述实施例2提供的cc@cu催化材料应用于(戊)
‑1‑
苯基
‑3‑
(4
‑
(三氟甲基)苯基)丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(戊)
‑1‑
苯基
‑3‑
(4
‑
(三氟甲基)苯基)丙
‑2‑
烯
‑1‑
酮为0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
5(66.0mg),收率为80%。
[0089][0090]
目标产物的核磁氢谱和碳谱如下所示:
[0091]1h nmr(400mhz,chloroform
‑
d);δ=7.82
–
7.80(m,2h),7.55
–
7.51(m,1h),7.44
–
7.34(m,9h),7.08(d,j=8hz,2h),3.56(dd,j=17.4,10.5hz,1h),3.28
–
3.18(m,2h),0.32(s,3h),0.27(s,3h).
[0092]
13
c nmr(100mhz,chloroform
‑
d);δ=198.5,147.0,136.8,136.0,134.1,133.1,129.6,128.6,128.0,127.9,127.71,127.14,126.8,125.8,125.10,125.06,125.0,124.99,123.1,38.5,31.5,
‑
4.0,
‑
5.2.
[0093]
应用例5表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(戊)
‑1‑
苯基
‑3‑
(4
‑
(三氟甲基)苯基)丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了80%。
[0094]
应用例6:
[0095]
将上述实施例2提供的cc@cu催化材料应用于(e)
‑3‑
(4
‑
甲氧基苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(e)
‑3‑
(4
‑
甲氧基苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
6(61.4mg),收率为82%。
[0096][0097]
目标产物的核磁氢谱和碳谱如下所示:
[0098]1h nmr(400mhz,chloroform
‑
d);δ=7.80
–
7.78(m,2h),7.52
–
7.45(m,3h),7.41
–
7.33(m,5h),6.93
–
6.88(m,2h),6.75
–
6.72(m,2h),3.75(s,3h),3.46(dd,j=17.0,10.5hz,1h),3.21
–
3.01(m,2h),0.30(s,3h),0.24(s,3h).
[0099]
13
c nmr(100mhz,chloroform
‑
d);δ=199.4,157.0,137.1,137.0,134.21,134.19,132.8,129.3,128.6,128.5,128.0,127.8,113.6,55.2,39.2,30.1,
‑
3.8,
‑
5.2.
[0100]
应用例6表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(e)
‑3‑
(4
‑
甲氧基苯基)
‑1‑
苯基丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了82%。
[0101]
应用例7:
[0102]
将上述实施例2提供的cc@cu催化材料应用于(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
7(48.7mg),收率为62%。
[0103][0104]
目标产物的核磁氢谱和碳谱如下所示:
[0105]1h nmr(400mhz,chloroform
‑
d);δ=7.70
–
7.68(m,2h),7.44
–
7.33(m,5h),7.20
–
7.18(m,2h),7.14
–
7.10(m,2h),6.90
–
6.86(m,2h),3.44(dd,j=16.9,10.4hz,1h),3.19
–
3.04(m,2h),2.38(s,3h),0.29(s,3h),0.23(s,3h).
[0106]
13
c nmr(100mhz,chloroform
‑
d);δ=198.4,143.8,141.1,136.4,134.4,134.2,130.3,129.5,129.2,128.9,128.2,128.1,127.9,38.5,30.8,21.7,
‑
4.0,
‑
5.2.
[0107]
应用例7表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了62%。
[0108]
应用例8:
[0109]
将上述实施例2提供的cc@cu催化材料应用于(戊)
‑3‑
(4
‑
氟苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(戊)
‑3‑
(4
‑
氟苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
8(51.2mg),收率为68%。
[0110][0111]
目标产物的核磁氢谱和碳谱如下所示:
[0112]1h nmr(400mhz,chloroform
‑
d);δ=7.70
–
7.67(m,2h),7.43
–
7.32(m,5h),7.19(d,j=8.1hz,2h),6.91
–
6.82(m,4h),6.90
–
6.86(m,2h),3.43(dd,j=17.0,10.6hz,1h),3.19
–
3.03(m,2h),2.38(s,3h),0.29(s,3h),0.24(s,3h).
[0113]
13
c nmr(100mhz,chloroform
‑
d);δ=198.6,161.8,159.3,143.7,138.0,136.6,
134.5,134.2,129.4,129.2,128.9,128.8,128.1,127.8,115.0,114.8,38.8,30.4,21.6,
‑
4.0,
‑
5.1.
[0114]
应用例8表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(戊)
‑3‑
(4
‑
氟苯基)
‑1‑
(对甲苯基)丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了68%。
[0115]
应用例9:
[0116]
将上述实施例2提供的cc@cu催化材料应用于(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(4
‑
氟苯基)丙
‑2‑
烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(4
‑
氟苯基)丙
‑2‑
烯
‑1‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
9(57.2mg),收率为72%。
[0117][0118]
目标产物的核磁氢谱和碳谱如下所示:
[0119]1h nmr(400mhz,chloroform
‑
d);δ=7.82
–
7.78(m,2h),7.44
–
7.33(m,5h),7.15
–
7.12(m,2h),7.08
–
7.03(m,2h),6.95
–
6.86(m,2h),3.42(dd,j=17.1,10.6hz,1h),3.18
–
3.02(m,2h),0.29(s,3h),0.25(s,3h).
[0120]
13
c nmr(100mhz,chloroform
‑
d);δ=197.2,166.9,164.3,140.9,136.3,134.1,133.30,133.28,130.6,130.51,130.46,129.5,128.8,128.3,127.9,115.7,115.5,38.7,30.8,
‑
4.0,
‑
5.3.
[0121]
应用例9表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(戊)
‑3‑
(4
‑
氯苯基)
‑1‑
(4
‑
氟苯基)丙
‑2‑
烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了72%。
[0122]
应用例10:
[0123]
将上述实施例2提供的cc@cu催化材料应用于反式
‑1‑
苯基
‑2‑
丁烯
‑1‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,反式
‑1‑
苯基
‑2‑
丁烯
‑1‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
10(42.4mg),收率为75%。
[0124][0125]
目标产物的核磁氢谱和碳谱如下所示:
[0126]1h nmr(400mhz,chloroform
‑
d);δ=7.83
–
7.81(m,2h),7.55
–
7.51(m,3h),7.43
–
7.37(m,5h),3.02(dd,j=15.8,3.3hz,1h),2.68
–
2.61(m,1h),1.63
–
1.59(m,1h),0.98(d,j=7.3hz,3h),0.33(d,j=3.3hz,6h).
[0127]
13
c nmr(100mhz,chloroform
‑
d);δ=200.7,137.6,137.1,134.0,132.8,129.2,128.5,128.1,127.9,40.7,15.9,14.6,
‑
4.7,
‑
5.4.
[0128]
应用例10表明,在本发明实施例提供的cc@cu催化材料的催化条件下,反式
‑1‑
苯基
‑2‑
丁烯
‑1‑
酮的转化率也很高,其硅加成产物的收率达到了75%。
[0129]
应用例11:
[0130]
将上述实施例2提供的cc@cu催化材料应用于反式苯亚甲基丙酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,反式苯亚甲基丙酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
11(45.2mg),收率为80%。
[0131][0132]
目标产物的核磁氢谱和碳谱如下所示:
[0133]1h nmr(400mhz,chloroform
‑
d);δ=7.42
–
7.32(m,5h),7.22
–
7.17(m,2h),7.11
–
7.06(m,1h),6.95
–
6.93(m,2h),2.96
–
2.87(m,2h),2.68
–
2.59(m,1h),1.95(s,3h),0.24(s,3h)),0.22(s,3h).
[0134]
13
c nmr(100mhz,chloroform
‑
d);δ=208.3,142.0,136.6,134.2,129.4,128.2,127.8,127.6,124.9,44.0,31.4,30.0,
‑
4.0,
‑
5.4.
[0135]
应用例11表明,在本发明实施例提供的cc@cu催化材料的催化条件下,反式苯亚甲基丙酮的转化率也很高,其硅加成产物的收率达到了80%。
[0136]
应用例12:
[0137]
将上述实施例2提供的cc@cu催化材料应用于(e)
‑3‑
戊烯
‑2‑
酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,(e)
‑3‑
戊烯
‑2‑
酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
12(34.4mg),收率为78%。
[0138][0139]
目标产物的核磁氢谱和碳谱如下所示:
[0140]1h nmr(400mhz,chloroform
‑
d);δ=7.51
–
7.48(m,2h),7.38
–
7.35(m,3h),2.44
–
2.39(m,1h),2.21
–
2.14(m,1h),2.07(s,3h),1.54
–
1.45(m,1h),0.93(d,j=7.3hz),0.27
(s,6h).
[0141]
13
c nmr(100mhz,chloroform
‑
d);δ=209.5,137.5,133.9,129.1,127.8,45.9,30.0,15.2,14.5,
‑
4.8,
‑
5.3.
[0142]
应用例12表明,在本发明实施例提供的cc@cu催化材料的催化条件下,(e)
‑3‑
戊烯
‑2‑
酮的转化率也很高,其硅加成产物的收率达到了78%。
[0143]
应用例13:
[0144]
将上述实施例2提供的cc@cu催化材料应用于反式肉桂醛与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,反式肉桂醛0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,从而得硅加成产物,反应结束后,过滤整个反应体系,以乙酸乙酯10ml洗涤,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=9:1柱层析纯化得到有机硅化合物ii
‑
13(40.3mg),收率为75%。
[0145][0146]
目标产物的核磁氢谱和碳谱如下所示:
[0147]1h nmr(400mhz,chloroform
‑
d);δ=9.54
–
9.53(m,1h),7.42
–
7.33(m,5h),7.23
–
7.19(m,2h),7.13
–
7.09(m,1h),6.96
–
6.94(m,2h),2.91
–
2.83(m,2h),2.67
–
2.59(m,1h),0.27(d,j=11.4hz,6h).
[0148]
13
c nmr(100mhz,chloroform
‑
d);δ=202.7,141.1,136.2,134.1,129.5,128.3,127.9,127.7,125.2,43.5,30.1,
‑
4.2,
‑
5.5.
[0149]
应用例13表明,在本发明实施例提供的cc@cu催化材料的催化条件下,反式肉桂醛的转化率也很高,其硅加成产物的收率达到了75%。
[0150]
应用例14:
[0151]
将上述实施例2提供的cc@cu催化材料应用于查尔酮与联硼酸频那醇二甲基硅试剂的硅加成反应,其中,查尔酮0.20mmol,联硼酸频那醇二甲基硅试剂为0.24mmol,催化材料为0.002mmol,水为1.0ml,室温反应时间为12h,反应结束后,过滤整个反应体系,以乙酸3ml洗涤。向残留物中直接加入溴化钾24mg,过乙酸30mg,整个体系在室温下搅拌12小时,以乙酸乙酯10ml稀释反应体系,再以乙酸乙酯(3
×
10ml)萃取,分离出有机相后,用无水na2so4干燥,过滤,旋转蒸发除去溶剂。残留物经乙酸乙酯/石油醚混合溶剂=4:1柱层析纯化得到产率82%。。
[0152][0153]
应用例14表明,在本发明实施例提供的cc@cu催化材料的催化条件下,查尔酮可使用“一锅法”制备β
‑
羟基化合物,其转化产物的收率达到了82%。
[0154]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和
原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1