含螺双茚并双苯并噁唑聚酰亚胺气体分离膜及其制备方法
1.本技术涉及有机合成技术和气体分离领域,具体来说,本技术涉及一种含螺双茚并双苯并噁唑聚酰亚胺气体分离膜以及该含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的制备方法。
背景技术:
2.自21世纪以来,随着全球人口和经济的高速发展,能源和环境问题日益突出。膜基气体分离技术已经在油气开采、大气环境管理和其它化学领域受到许多关注。
3.已证实包括螺双茚骨架结构的聚合物膜材料是改善气体分离性能、热稳定性以及溶解度的行之有效的方法。螺结构由通过特定正四面体键接原子正交地连接的两个环组成。通常碳原子作为螺心。连接螺环结构的两个环各自是平坦的,但它们彼此垂直。螺链段的这种结构特征迫使聚合物链在每个螺环中心处以90℃的角度扭曲,降低了聚合物链之间相互作用的概率,并带来更高的聚合物气体分离性能和溶解度。此外,螺片段还能保持聚合物链的刚性,确保聚合物的热稳定性。近年来已报道了许多包含螺双茚的聚酰亚胺气体分离膜,螺双茚结构主要包括在二胺中或者包括在二酐中。这些气体分离膜具有优异的透过性和选择性。
4.近年来,已广泛利用将α-羟基聚酰亚胺前驱体高温转化成聚苯并噁唑(pbo)来制备高性能的气体分离膜,即所谓的高温重排膜。与其它聚合物膜相比,因为热驱动的固体状态时的结构重排在膜中形成了特定的微孔和孔径分布,高温重排膜展现出优异的气体分离性能。但是,与常规的聚酰亚胺薄膜相比,有些含螺双茚的聚酰亚胺气体分离膜的机械性能较差,且当α-羟基聚酰亚胺前驱体在大于400℃的高温下处理并发送交联后,它们是不溶的。此外,当在450℃的高温下处理α-羟基聚酰亚胺前驱体时,它们可能发生链分解。
5.为此,本领域持续需要开发一种机械性能好的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜及其制备方法。
技术实现要素:
6.本技术之目的在于提供具有新结构的螺双茚并双苯并恶唑二胺,并以其作为二胺单体来制备具有良好机械性能的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜,从而解决上述技术问题。
7.本技术之目的还在于提供一种含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的制备方法。
8.为了解决上述技术问题,本技术提供以下技术方案。
9.在第一方面中,本技术提供一种含螺双茚并双苯并噁唑聚酰亚胺气体分离膜,其特征在于,其具有通过下述通式(1)所示的结构:
[0010][0011]
在上述通式(1)中,m为大于0的正整数,n为0或者大于或等于1的正整数;
[0012]
ar为螺双茚并双苯并恶唑二胺,所述螺双茚并双苯并恶唑二胺具有通过下述结构式i、结构式ii、结构式iii或者结构式iv所示的结构;
[0013]
其中,结构式i如下所示:
[0014][0015]
其中,结构式ii如下所示:
[0016][0017]
其中,结构式iii如下所示:
[0018][0019]
以及,其中结构式iv如下所示:
[0020][0021]
在第一方面的一种实施方式中,m和n的比值为1:1。
[0022]
在第一方面的一种实施方式中,所述含螺双茚并双苯并噁唑聚酰亚胺气体分离膜中聚酰亚胺的数均分子量可在35000-110000之间,重均分子量可在7200-232000之间,分子量分布系数可在1.7-2.5之间。例如,数均分子量可为35000、40000、45000、50000、60000、
70000、80000、90000、100000、110000或者它们中任意两个数值之间的范围或者子范围。分子量分布系数可为1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5或者它们中任意两个数值之间的范围或子范围。
[0023]
在第一方面的一种实施方式中,所述含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的厚度为30-50微米。
[0024]
在第二方面中,本技术提供一种如第一方面所述的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的制备方法,其特征在于,所述方法包括以下步骤:
[0025]
s1:在室温下,将螺双茚并双苯并恶唑二胺、4,4-六氟异丙基邻苯二甲酸酐和任选的对苯二胺溶于第一有机溶剂中,得到聚酰胺酸溶液;
[0026]
s2:对所述聚酰亚胺酸溶液进行化学亚胺化,得到聚酰亚胺固体;
[0027]
s3:将所述聚酰亚胺固体溶于第二有机溶剂中,得到聚酰亚胺溶液,完全溶解后过滤,浇注到玻璃皿上,在干燥的环境氛围下自然挥发24小时,然后在80-150℃的温度下真空干燥,得到所述含螺双茚并双苯并噁唑聚酰亚胺气体分离膜。在该实施方式中,采用化学亚胺化,并在玻璃皿中自然挥发成膜,再进一步加热真空除溶剂,其最主要目的是为了得到的膜pi分子链堆积更松散,聚合物自由体积大,气体透过性好,故采用化学亚胺化,常温成膜。
[0028]
在第二方面的一种实施方式中,所述第一有机溶剂为n,n-二甲基乙酰胺或者n,n-二乙基乙酰胺;所述第二有机溶剂为氯仿。
[0029]
在第二方面的一种实施方式中,在步骤s2中,对所述聚酰胺酸溶液进行化学亚胺化包括将乙酸酐和吡啶的混合溶液缓慢加入聚酰胺酸溶液中,然后在120℃加热8小时以形成聚酰亚胺。
[0030]
在第二方面的一种实施方式中,在步骤s3中,所述聚酰亚胺固体溶液中聚酰亚胺的质量分数为10-20%。
[0031]
在第二方面的一种实施方式中,所述螺双茚并双苯并恶唑二胺通过以下步骤来制备:
[0032]
在存在脱水剂的惰性气氛下,且在180-220℃的反应温度下,使起始化合物与氨基取代的苯甲酸在质子酸溶剂中反应第一预定时间段,得到螺双茚并双苯并恶唑二胺;
[0033]
其中,所述起始化合物具有通过下述结构式v或者结构式vi所示的结构,其中,所述结构式v如下所示:
[0034][0035]
其中,所述结构式vi如下所示:
[0036][0037]
其中,所述氨基取代的苯甲酸为3-氨基苯甲酸或者4-氨基苯甲酸。
[0038]
在第二方面中,所述脱水剂为五氧化二磷。所述质子酸溶剂为多聚磷酸。所述第一预定时间段为8-12小时。
[0039]
与现有技术相比,本技术的有益效果在于利用本文所述的含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜又具有特定结构的螺双茚并双苯并恶唑二胺制成,具备优异的耐热性、优秀的力学性能、溶解性和透过性以及选择性。
附图说明
[0040]
图1显示根据实施例1的螺双茚并双苯并恶唑二胺的核磁氢谱图谱。
[0041]
图2显示根据实施例2的螺双茚并双苯并恶唑二胺的核磁氢谱图谱。
[0042]
图3显示根据实施例3的螺双茚并双苯并恶唑二胺的核磁氢谱图谱。
[0043]
图4显示根据实施例4的螺双茚并双苯并恶唑二胺的核磁氢谱图谱。
[0044]
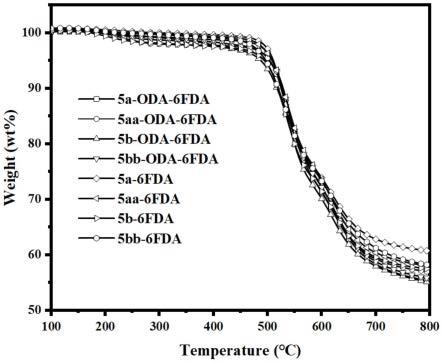
图5为根据实施例5-12制备的含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜的热重分析图。
[0045]
图6为根据实施例5-12制备的含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜的动态机械分析图。
[0046]
图7显示根据本发明的一种实施方式的螺双茚并双苯并恶唑二胺的合成路线。
[0047]
图8显示根据本发明的一种实施方式的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的合成路线。
具体实施方式
[0048]
如上所述,聚酰亚胺气体分离膜日益受到关注,但现有技术中的含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜存在机械性能和耐热性不强导致不能在极端条件下稳定使用以及气体高透过选择性等问题。为此,本技术提供一种耐热性优异、机械性能优秀以及良好的气体透过选择性的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜。
[0049]
在一种实施方式中,本技术提供一种含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜,其特征在于,其具有通过下述通式(1)所示的结构:
[0050][0051]
在上述通式(1)中,m为大于0的正整数,n为0或者大于或等于1的正整数;
[0052]
ar为螺双茚并双苯并恶唑二胺,所述螺双茚并双苯并恶唑二胺具有通过下述结构式i、结构式ii、结构式iii或者结构式iv所示的结构;
[0053]
其中,结构式i如下所示:
[0054]
[0055]
其中,结构式ii如下所示:
[0056][0057]
其中,结构式iii如下所示:
[0058][0059]
以及,其中结构式iv如下所示:
[0060][0061]
在一种具体实施方式中,m和n的比值为1:1。在一种优选的实施方式中,n的数值可为0。换句话说,可以不使用对苯二胺作为二胺单体来合成聚酰亚胺。
[0062]
在一种具体实施方式中,结构式i、结构式ii、结构式iii或者结构式iv所示螺双茚并双苯并恶唑二胺的化学名称分别为4,4'-(5,5,5',5'-四甲基-5,5',6,6'-四氢-7,7'-螺环双[茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺、3,3'-(5,5,5',5'-四甲基-5,5',6,6'-四氢-7,7'-螺环双[茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺、4,4'-(5',5',6,6-四甲基-5',6,6',7-四氢螺环[茚并[4,5-d]噁唑-8,7'-茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺,以及3,3'-(5',5',6,6-四甲基-5',6,6',7-四氢螺环[茚并[4,5-d]噁唑-8,7'-茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺。
[0063]
在另一种实施方式中,本技术提供一种含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的制备方法。在一种具体实施方式中,所述方法可包括以下步骤:
[0064]
s1:在室温下,将螺双茚并双苯并恶唑二胺、4,4-六氟异丙基邻苯二甲酸酐和任选的对苯二胺溶于第一有机溶剂中,得到聚酰胺酸溶液;
[0065]
s2:对所述聚酰亚胺酸溶液进行化学亚胺化,得到聚酰亚胺固体;
[0066]
s3:将所述聚酰亚胺固体溶于第二有机溶剂中,得到聚酰亚胺溶液,完全溶解后过滤,浇注到玻璃皿上,在干燥的环境氛围下自然挥发24小时,然后在80-150℃的温度下真空干燥得到所述含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜。
[0067]
在一种优选的实施方式中,本技术提供一种螺双茚并双苯并恶唑二胺的制备方法,所述方法包括在存在脱水剂的惰性气氛下,且在180-220℃的反应温度下,使起始化合物与氨基取代的苯甲酸在质子酸溶剂中反应第一预定时间段,得到螺双茚并双苯并噁唑环二胺;
[0068]
其中,所述起始化合物具有通过下述结构式v或者结构式vi所示的结构,其中,所述结构式v如下所示:
[0069][0070]
其中,所述结构式vi如下所示:
[0071][0072]
其中,所述氨基取代的苯甲酸为3-氨基苯甲酸或者4-氨基苯甲酸。
[0073]
在一种具体实施方式中,所述脱水剂为五氧化二磷。在一种具体实施方式中,所述质子酸溶剂为多聚磷酸。在一种具体实施方式中,所述第一预定时间段为8-12小时。在一种优选的实施方式中,反应温度可为180℃、190℃、200℃、210℃、220℃或者它们中任意两个数值之间的范围或子范围。在一种优选的实施方式中,所述第一预定时间段为8小时、8.5小时、9小时、9.5小时、10小时、10.5小时、11小时、11.5小时、12小时或者它们中任意两个数值之间的范围或者子范围。
[0074]
发明人首次合成了如上所述的螺双茚并双苯并恶唑二胺,并通过核磁图谱以及红外图谱等对其进行了表征,通过对图谱进行分析,可以确认成功制备了如上所述的螺双茚并双苯并恶唑二胺。在一种实施方式中,本文所述的螺双茚并双苯并恶唑二胺可与酸酐单体发生缩聚反应得到聚酰胺酸,通过亚胺化特别是化学亚胺化之后,可以得到聚酰亚胺固体。以聚酰亚胺固体作为原料可制备含螺双茚并双苯并噁唑聚酰亚胺气体分离膜。
[0075]
在一种具体实施方式中,对于聚酰亚胺的制备,使用了传统的两步法(化学亚胺化)。在三颈烧瓶中,将螺双茚并双苯并恶唑二胺(5.0mmol)加入到15ml dmac(二甲基乙酰胺)中。在室温下在n2下将该混合物搅拌30分钟。将二酐(5.0mmol)加入该溶液中,搅拌过夜并保持在室温后,形成粘性聚酰胺酸。将乙酸酐(5.0ml)和吡啶(2.5ml)的混合溶液缓慢加入聚酰胺酸中,然后在120℃加热8小时以形成聚酰亚胺。将反应液倒入乙醇/水混合物(1:1,500ml)中;随后通过过滤收集聚合物,随后用乙醇索格利特萃取24小时并在120℃真空干燥12小时以提供呈白色固体状的聚酰亚胺(0.55g,产率:92%)。
[0076]
实施例
[0077]
下面将结合实施例,对本技术进行一步描述和说明。如无特别说明,所用化工原料均可从市场购买。本领域技术人员可以理解,下述实施例只是示例性的。
[0078]
在下述实施例中,所用表征方法如下所述。
[0079]
核磁共振氢谱(1h nmr):
[0080]
反应产物与中间体的核磁共振波谱(1h nmr)图谱在德国布鲁克avance iii hd 400/500上获得。制样方法为:在洁净干燥的玻璃核磁管中,将10mg左右样品完全溶解于0.5ml左右的氘代试剂中。较好溶解的产物以氘代氯仿做溶剂(cdcl3)室温下溶解,溶解性较差的产物以氘代二甲基亚砜(dmso-d6)做溶剂,dmso-d6在较低室温下容易凝固,上样前需用吹风机吹化。常温下测试时以四甲基硅烷(tms)作为内标,化学位移为0ppm。
[0081]
傅里叶红外变换光谱(ft-ir):
[0082]
在干燥的环境中,采用nicolet is5(thermofisher scientific,inc.,usa)傅立叶变换红外光谱仪(ftir)atr模式进行测试,测试前单体(4nada)先置于真空烘箱中真空80℃2h除去水分。扫描范围设为4000-650cm-1
,分辨率设定为2cm-1
,扫描次数设定为32次,自动取平均值。
[0083]
热性能分析(tga&dma&tma):
[0084]
聚酰亚胺气体分离膜的热失重行为在ta discovery 550型热重分析仪(tga)上,氮气保护下进行,气体流量为50ml/min,以20℃/min从室温上升至120℃停留15min后,在以10℃/min从50℃上升至800℃。
[0085]
玻璃化转变温度(tg)在ta q800动态热机械分析仪(dma)上进行,将透明聚酰亚胺裁剪成宽度一样(0.53cm)的矩形长条,加载频率设置为1hz,以5℃/min的速率从30℃升至550℃,氮气保护。
[0086]
力学性能分析:
[0087]
聚酰亚胺气体分离膜的力学性能通过三思泰捷sust cmt1104电子万能试验机测量,试样裁剪为宽为1cm的矩形长条,牵引速率为5mm/min,遵照astm d882-02测试标准,取多次有效试验结果的平均值。
[0088]
气体分离性能:
[0089]
本技术通过实验室搭建的气体分离测试系统来测试本文所述的含螺双茚并双苯并噁唑的聚酰亚胺气体分离膜的气体分离性能。该气体分离测试系统及测试方法可参考xiaohua ma等发表在j.mater.chem.a,2021,9,5404的论文《unprecedented gas separation performance of a difluoro-functionalized triptycene-based ladder pim membrane at low temperature(低温下双氟功能化三联烯基梯形pim膜的前所未有的气体分离性能)》。
[0090]
气体渗透率由恒容/变压时滞法测定。低压室压力使用inficon传感器记录,范围为0-10torr;仪器的进气速率为~1
×
10-7
torr/s,低压室容积为30cm3。在测试之前,使用聚砜气体分离膜作为标准膜材料来校准系统。在35℃下获得的o2渗透率为1.25barrer,o2/n2选择性为5.5,这与之前报告的数据非常吻合。聚合物薄膜在测试前脱气24小时,气体透过率在2barrer高压室压力下使用以下公式:
[0091][0092]
其中p是渗透率(barrer):1barrer=10-10
cm3(stp)cm cm-2
s-1
cmhg-1
,vd是校准渗透体积(cm3),ι是膜厚度(cm),p
up
高压室的压强(cmhg),a是有效膜面积(cm2),t是工作温度(k),r是气体常数(r=0.278cm3cmhg cm-3
(stp)k-1
),dp/dt是稳态下低压侧压力随时间的变
化率(cmhgs-1
)。各向同性聚合物膜的表面扩散系数d(cm2s-1
)使用d=ι2/6θ计算,其中ι是膜厚度,θ是渗透率测量的时间滞后。溶解度系数s(cm3(stp)cm-3
cmhg-1
)由关系式s=p/d获得。
[0093]
气体选择性(α
a/b
)可以表示为:α
a/b
=pa/pb。对八种螺双茚并双苯并噁唑聚酰亚胺膜进行了测试,渗透率和选择性值的偏差在
±
5%的范围内。
[0094]
单体合成实施例
[0095]
参考图7,在下述实施例中,具有结构式v和具有结构式vi的起始化合物可通过以下步骤来合成。
[0096]
第一步:合成3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-6,6
′‑
二醇。
[0097]
将双酚a(100g,0.439mol)和甲磺酸(10ml)在反应烧瓶中混合并加热至135℃,保持4小时。在剧烈搅拌下将所得棕色粘性油倒入水(2000ml)中。最后用乙醇/水混合溶剂重结晶,得到白色针状结晶(25.0g,收率55.5%)。(tlc:二氯甲烷/石油醚=1/4,rf=0.25)。
[0098]
第二步:合成3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二硝基-6,6
′‑
二醇以及3,3,3',3'-四甲基-5,7'-二硝基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇。
[0099]
将3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-6,6
′‑
二醇(3.08g,10.0mmol)溶解于乙酸(100ml)中,滴加hno3(4n,2.1当量,5.3ml)和乙酸(50.0ml)的混合溶液。将混合浆液搅拌过夜,然后在过滤前冷却至2-5℃。通过柱分离得到淡黄色固体3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二硝基-6,6
′‑
二醇(tlc:二氯甲烷/石油醚=1/1,rf=0.6)和3,3,3',3'-四甲基-5,7'-二硝基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇(tlc:二氯甲烷/石油醚=1/1,rf=0.7)。
[0100]
第三步:合成3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二氨基-6,6
′‑
二醇以及5,7'-二氨基-3,3,3',3'-四甲基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇。
[0101]
将黄色固体3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二硝基-6,6
′‑
二醇或3,3,3',3'-四甲基-5,7'-二硝基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇(10g,25.0mmol)分散在乙醇(200ml)中,然后与加热回流的pd/c(0.8g)混合。然后,将n2h4·
h2o(80%,20ml)滴加到热溶液中。回流10h后,过滤除去沉淀,将溶液冷却至室温,加入200ml水,收集白色沉淀。固体在乙醇中重结晶并真空干燥,最终得到白色粉末3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二氨基-6,6
′‑
二醇或5,7'-二氨基-3,3,3',3'-四甲基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇(9.4g,96%)。
[0102]
实施例1
[0103]
本实施例涉及合成具有结构式i的螺双茚并双苯并恶唑二胺,即4,4'-(5,5,5',5'-四甲基-5,5',6,6'-四氢-7,7'-螺环双[茚并[5,6-d]噁唑]-2,2'-二基)双苯胺(5a)。
[0104]
本实施例的具体合成步骤如下所述。
[0105]
将多聚磷酸(160g)和p2o5(20g)放入一个完全干燥的500ml三颈烧瓶中,该烧瓶配备有机械搅拌器和氮气入口/出口。将混合物在100℃搅拌和加热,直到p2o5完全溶解。冷却至室温后,将3,3,3,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二氨基-6,6
′‑
二醇(13.5g,40.0mmol)和3-氨基苯甲酸(11.6g,84.6mmol)搅拌到混合物中以产生稠糊。将所得混合物缓慢加热至200℃并在此温度下保持10小时。冷却至100℃后,将反应混合物倒入冰冷的水中,快速搅拌。过滤收集沉淀,然后在5%碳酸氢钠溶液中浸泡过夜。粗产物通过中性氧化铝色谱纯化,用乙酸乙酯/石油醚(3:1,v/v,rf=0.6)洗脱,得到白色固体(17.9g,83%)。
[0106]
对根据实施例1制备的螺双茚并双苯并恶唑二胺进行核磁氢谱表征和红外图谱表征。该螺双茚并双苯并恶唑二胺的红外解析数据如下:ftir(kbr,cm-1
):3458,3375,3322,3192(胺nh),2950,2925(ch3),2860(ch2),1605(nh2,c=n),1173(噁唑c-o-c)。该螺双茚并双苯并恶唑二胺的核磁氢谱如图1所示,具体解析数据如下:1h nmr(dmso-d6,600mhz):δ7.79(d,j=7.8hz,4h,ar-h),7.48(s,2h,ar-h),6.89(s,2h,ar-h),6.67(d,j=7.8hz,4h,ar-h),5.94(s,4h,nh2),2.42(s,2h,ch2),2.42(s,2h,ch2),2.28(s,2h,ch2),1.42(s,6h,ch3),1.33(s,6h,ch3)ppm.
13
c nmr(dmso-d6,150mhz):δ164.1,152.8,150.2,149.1,147.8,142.5,129.2,114.0,113.6,112.1,105.7,60.2,60.0,57.6,32.3,30.8ppm。
[0107]
实施例2
[0108]
本实施例涉及合成具有结构式ii的螺双茚并双苯并恶唑二胺,即3,3'-(5,5,5',5'-四甲基-5,5',6,6'-四氢-7,7'-螺环双[茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺(5aa)。
[0109]
该实施例的合成步骤与实施例1相似,但将原料替换成将3,3,3
′
,3
′‑
四甲基-1,1
′‑
螺环双茚满-5,5
′‑
二氨基-6,6
′‑
二醇(13.5g,40.0mmol)和4-氨基苯甲酸(11.6g,84.6mmol)。最终得到白色固体(17.5g,81%)。
[0110]
对根据实施例2制备的螺双茚并双苯并恶唑二胺进行核磁氢谱表征和红外图谱表征。该螺双茚并双苯并恶唑二胺的红外解析数据如下:ftir(kbr,cm-1
):3406,3317,3207(胺nh),2950,2925(ch3),2860(ch2),1605(nh2,c=n),1173(噁唑c-o-c)。该螺双茚并双苯并恶唑二胺的核磁氢谱如图2所示,具体解析数据如下:1h nmr(dmso-d6,600mhz):δ7.63(s,2h,ar-h),7.36(s,2h,ar-h),7.25(d,j=7.8hz,2h,ar-h),7.19(dd,j=7.8hz,2h,ar-h),7.04(s,2h,ar-h),6.76(d,j=7.8hz,2h,ar-h),5.47(s,4h,nh2),2.48(d,j=12.6hz,2h,ch2),2.34(d,j=13.2hz,2h,ch2),1.48(s,6h,ch3),1.39(s,6h,ch3)ppm.
13
c nmr(dmso-d6,150mhz):δ163.6,150.5,149.8,149.6,149.0,142.1,130.1,127.61,117.58,114.9,113.1,112.3,106.2,60.23,60.0,57.7,32.3,30.8ppm。
[0111]
实施例3
[0112]
本实施例涉及合成具有结构式iii的螺双茚并双苯并恶唑二胺,即4,4'-(5',5',6,6-四甲基-5',6,6',7-四氢螺环[茚并[4,5-d]噁唑-8,7'-茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺(5b)。
[0113]
该实施例的合成步骤与实施例1相似,但将原料替换成将5,7'-二氨基-3,3,3',3'-四甲基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇(13.5g,40.0mmol)和3-氨基苯甲酸(11.6g,84.6mmol)。最终得到白色固体(16.4g,76%)。
[0114]
对根据实施例3制备的螺双茚并双苯并恶唑二胺进行核磁氢谱表征和红外图谱表征。该螺双茚并双苯并恶唑二胺的红外解析数据如下:ftir(kbr,cm-1
):3468,3342,3223(胺nh),2950,2925(ch3),2860(ch2),1605(nh2,c=n),1173(噁唑c-o-c)。该螺双茚并双苯并恶唑二胺的核磁氢谱如图3所示,具体解析数据如下:1h nmr(dmso-d6,600mhz):δ7.74(d,j=9.0hz,2h,ar-h),7.58(d,j=8.4hz,2h,ar-h),7.54(d,j=8.4hz,1h,ar-h),7.51(s,1h,ar-h),7.20(d,j=8.4hz,1h,ar-h),6.78(s,1h,ar-h),6.64(d,j=9.0hz,2h,ar-h),6.57(d,j=9.0hz,2h,ar-h),5.90(s,2h,nh2),5.87(s,2h,nh2),2.98(d,j=12.6hz,1h,ch2),2.47(d,j=13.2hz,1h,ch2),2.32(d,j=13.2hz,1h,ch2),2.80(d,j=13.2hz,1h,ch2),
1.57(s,3h,ch3),1.45(s,3h,ch3),1.40(s,3h,ch3),1.34(s,3h,ch3)ppm.
13
c nmr(dmso-d6,150mhz):δ163.9,163.6,152.7,150.7,150.0,148.8,148.6,148.0,142.2,138.5,138.3,129.2,129.1,118.0,114.0,113.7,113.4,112.3,112.3,109.8,105.0,61.0,57.5,56.9,32.8,32.3,31.3,30.7ppm。
[0115]
实施例4
[0116]
本实施例涉及合成具有结构式iv的螺双茚并双苯并恶唑二胺,即3,3'-(5',5',6,6-四甲基-5',6,6',7-四氢螺环[茚并[4,5-d]噁唑-8,7'-茚并[5,6-d]噁唑]-2,2'-二取代基)双苯胺(5bb)。
[0117]
该实施例的合成步骤与实施例1相似,但将原料替换成将5,7'-二氨基-3,3,3',3'-四甲基-2,2',3,3'-四氢-1,1'-螺环双[茚]-6,6'-二醇(13.5g,40.0mmol)和4-氨基苯甲酸(11.6g,84.6mmol)。最终得到白色固体(17.1g,79%)。
[0118]
对根据实施例4制备的螺双茚并双苯并恶唑二胺进行核磁氢谱表征和红外图谱表征。该螺双茚并双苯并恶唑二胺的红外解析数据如下:ftir(kbr,cm-1
):3448,3334,3224(胺nh),2950,2925(ch3),2860(ch2),1605(nh2,c=n),1173(噁唑c-o-c)。该螺双茚并双苯并恶唑二胺的核磁氢谱如图4所示,具体解析数据如下:1h nmr(dmso-d6,600mhz):δ7.19(dd,j=3.6hz,2h,ar-h),7.33(s,1h,ar-h),7.31(d,j=8.4hz,1h,ar-h),7.22(d,j=7.8hz,1h,ar-h),7.15-7.17(m,1h,ar-h),7.06-7.11(m,1h,ar-h),6.90(s,1h,ar-h),6.73(dd,j1=7.8hz,j2=1.2hz,1h,ar-h),6.68(d,j=7.8hz,1h,ar-h),5.43(s,2h,nh2),5.39(s,2h,nh2),3.00(d,j=12.6hz,1h,ch2),2.51(d,j=13.2hz,1h,ch2),2.32-2.36(m,2h,ch2),1.60(s,3h,ch3),1.47(s,3h,ch3),1.42(s,3h,ch3),1.35(s,3h,ch3)ppm.
13
c nmr(dmso-d6,150mhz):δ163.5,163.2,151.1,149.9,149.8,149.4,149.2,141.1,139.2,138.0,130.2,127.8,127.6,119.5,117.7,117.6,115.1,115.0,113.4,112.4,112.3,110.5,105.6,61.1,57.7,57.2,32.8,32.4,31.4,30.9ppm。
[0119]
聚酰亚胺气体分离膜制备实施例
[0120]
参考图8,在下述实施例5-12中,通过以下步骤来制备含螺双茚并双苯并噁唑聚酰亚胺气体分离膜。
[0121]
本技术使用传统的两步法(化学亚胺化)来制备聚酰亚胺。
[0122]
首先,制备聚酰胺酸溶液。具体步骤如下:在三颈烧瓶中,将螺环双苯并噁唑二胺(2.5或5.0mmol)和4,4'-二氨基二苯醚(2.5或0mmol)加入到15ml dmac(二甲基乙酰胺)中,在室温下且在n2下将该混合物搅拌30分钟。然后,将二酐单体(5.0mmol)加入该溶液中,搅拌过夜并保持在室温后,形成粘性聚酰胺酸。在包括两种二胺单体的情况下,每种二胺单体的用量各位2.5mmol。
[0123]
其次,制备聚酰亚胺。具体步骤如下:将乙酸酐(5.0ml)和吡啶(2.5ml)的混合溶液缓慢加入聚酰胺酸中,然后在120℃加热8小时以形成聚酰亚胺。将反应液倒入乙醇/水混合物(1:1,500ml)中,随后通过过滤收集聚合物,随后用乙醇索格利特萃取24小时并在120℃真空干燥12小时以提供呈白色固体状的pi(0.55g,产率:92%)。
[0124]
将聚酰亚胺固体溶在氯仿溶液中(质量分数约10wt%),完全溶解后通过0.25μm pfte过滤器过滤以去除不溶性杂质。随后将该溶液浇铸到玻璃培养皿上,将其放在干燥氛围中室温下缓慢挥发24小时,将薄膜分别在80℃和150℃下真空烘箱分别干燥2小时将溶剂
除尽。最后通过浸入热水将薄膜从基材上除去。薄膜厚度约为30-50微米。
[0125]
将根据实施例5-12制备的聚酰亚胺薄膜分别命名为pi-1至pi-8。根据实施例5-12的聚酰亚胺薄膜的单体组成和分子量性能如表1所示。
[0126]
表1:根据实施例5-12的聚酰亚胺气体分离膜的单体组成和分子量
[0127][0128]
此外,分别测定了根据实施例5-12的聚酰亚胺薄膜的溶解性能、热学性能、力学性能、光学性能以及气体分离性能。结果分别如下表2-表6所示。
[0129]
表2:根据实施例5-12的聚酰亚胺薄膜的溶解性能
[0130][0131][0132]
表3:根据实施例5-12的聚酰亚胺薄膜的热学和力学性能
[0133][0134]
表4:根据实施例5-12的聚酰亚胺薄膜的气体分离性能
[0135][0136]
从表1可知,本文所述的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜的分子量和分子量分布系数可调节。例如,数均分子量可在35000-110000之间,重均分子量可在7200-232000之间,分子量分布系数可在1.7-2.5之间。
[0137]
从表2可知,这些聚酰亚胺具有很好的溶解度。可归因于螺环庞大和扭曲的非共面结构。使得聚酰亚胺分子链堆积松散,减弱了分子间和分子内的相互作用。观察到聚酰亚胺易溶于dmac、dmf、dmso、nmp、dcm、chcl3、thf,表明它们具有良好的溶液加工性。
[0138]
从表3可知,由于螺双茚并双苯并恶唑的高度扭曲和刚性结构,可以很好的限制聚酰亚胺分子链运动,导致聚酰亚胺高tg的原因。同时有良好热稳定性和机械性能。
[0139]
从表4可知,本文所述的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜具有良好的透过性和选择性。含螺双茚并双苯并噁唑聚酰亚胺气体分离膜具有良好的透过性和选择性归因于双茚并双苯并噁唑二胺结构的高度刚性和螺环结构的扭曲性导致聚酰亚胺分子链不能紧密堆积,聚合物自由体积大,从而避免形成致密的聚合物薄膜,为气体的透过和选择性提供了通道。
[0140]
总之,本文所述的含螺双茚并双苯并噁唑聚酰亚胺气体分离膜在具有优良的透过性和选择性的同时,还具备优异的热学和力学性能。
[0141]
上述对实施例的描述是为了便于本技术领域的普通技术人员能理解和应用本技术。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其它实施例中而不必付出创造性的劳动。因此,本技术不限于这里的实施例,本领域技术人员根据本技术披露的内容,在不脱离本技术范围和精神的情况下做出的改进和修改都本技术的范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1