一种还原催化剂和一种3-甲基-3,4-二氢-2H-1,4-苯并恶嗪的制备方法
一种还原催化剂和一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法
技术领域
1.本发明涉及农药中间体合成技术领域,尤其涉及一种还原催化剂和一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法。
背景技术:
2.除草剂安全剂是一种能够加强作物对除草剂的耐受能力、提高除草剂对作物的安全性的化合物。解草酮(n-二氯乙酰基-3-甲基-2h-1,4-苯并噁嗪)是一种二氯乙酰基苯并噁嗪类的除草剂安全剂,它能够提高作物的抗逆性,保护作物免受酰胺类除草剂的伤害,而被广泛用于田间,主要是保护玉米、高粱等作物免受甲草胺、异丙甲草胺等除草剂的危害,本身具有安全性高、使用方便等特点。
3.3-甲基-3,4-二氢-2h-1,4-苯并恶嗪是合成解草酮的重要中间体。目前,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪类化合物的合成方法一般有以下几种:(1)以邻硝基苯酚和3-溴丙烯为原料,经nah、钯催化氢化还原、合环制得(i las j et al.recent advances in the synthesis of 2h-1,4-benzoxazine-3-(4h)-ones and 3,4-dihydro-2h-1,4-benzoxazines.tetrahedron,2005,61(31):7325-7348.);(2)以对甲基苯磺酸和邻溴苯酚为原料,经nah、cui催化,还原制得(parai m k et al.,a convenient synthesis of chiral amino acid derived 3,4-dihydro-2h-benzothiazines and antibiotic levofloxacin.tetrahedr on lett,2009,50(33):4703-4705.);(3)以二氟硝基苯酚和甲基苯磺酸缩水甘油酯为原料,以钯碳作为催化剂在三苯膦和偶氮二甲酸二乙酯中催化还原并合环制得(苗华等.左氧氟沙星合成路线图解.中国医药工业杂志.1994,25(4):185-188.)。在上述合成3-甲基-3,4-二氢-2h-1,4-苯并恶嗪方法均使用了昂贵的催化剂,并且存在收率低的缺点。
技术实现要素:
4.有鉴于此,本发明提供了一种还原催化剂和一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法。本发明提供的催化剂成本低,催化效果好,利用该催化剂制备3-甲基-3,4-二氢-2h-1,4-苯并恶嗪时,产品的收率和纯度高。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.一种还原催化剂的制备方法,包括以下步骤:
7.将蛋膜粉与含有三价铁离子和二价钯离子的金属离子溶液进行第一混合,得到第一混合液;
8.将所述第一混合液和碱进行第二混合进行氢氧化反应,得到第二混合液;
9.将所述第二混合液和硼氢化盐溶液进行第三混合进行还原反应,得到第三混合液;
10.将所述第三混合液固液分离,得到固体前驱物;
11.将所述固体前驱物进行热处理,得到所述还原催化剂。
12.优选的,所述蛋膜粉与所述金属离子溶液的质量体积比为1g:(90~110)ml;所述金属离子溶液中三价铁离子的浓度为50~100mmol/l;所述金属离子溶液中二价钯离子的浓度为1.0~2.0mmol/l。
13.优选的,所述硼氢化盐溶液中硼氢根和三价铁离子的摩尔比为(0.1~0.5):1。
14.优选的,所述热处理在保护气氛下进行,所述热处理的温度为350~550℃,时间为3~4h,升温至所述热处理的温度的升温速率为3~7℃/min。
15.本发明还提供了上述技术方案所述制备方法制备得到的还原催化剂,包括载体和负载在载体上的铁单质和钯单质;所述载体为三维网状碳氮化合物。
16.本发明还提供了一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法,包括如下步骤:
17.将2-丙酮氧基硝基苯、还原催化剂和所述2-丙酮氧基硝基苯的良溶剂混合,在氢气气氛下进行还原-合环反应,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪;
18.所述还原催化剂为上述技术方案所述还原催化剂。
19.优选的,所述还原催化剂的质量为2-丙酮氧基硝基苯质量的20~50%;所述氢气的压力为0.2~0.5kpa;所述2-丙酮氧基硝基苯与所述2-丙酮氧基硝基苯的良溶剂的质量体积比为1g:(20~30)ml。
20.优选的,所述还原-合环反应的反应温度为30~50℃,反应时间为10~20h。
21.优选的,所述2-丙酮氧基硝基苯的良溶剂包括甲醇、乙醇、四氢呋喃、二氯甲烷和正丙醇中的任意一种或多种。
22.优选的,所述2-丙酮氧基硝基苯通过以下步骤制备得到:
23.将邻硝基苯酚、溴丙酮、无机碱性化合物、相转移催化剂和有机溶剂混合,进行取代反应,得到2-丙酮氧基硝基苯;
24.所述取代反应的反应温度为50~70℃,反应时间为4~8h;所述相转移催化剂为季铵盐;所述无机碱性化合物包括碱金属氢氧化物、碱金属碳酸盐和碱金属碳酸氢盐的任意一种或多种;所述有机溶剂包括甲苯、丙酮、乙腈、n,n-二甲基甲酰胺和二甲亚砜中的任意一种或多种;所述邻硝基苯酚、溴丙酮和无机碱性化合物的摩尔比为1:(0.5~2):(0.5~3);所述相转移催化剂的质量为邻硝基苯酚质量的3~10%;所述邻硝基苯酚与有机溶剂的质量体积比为1g:(2~5)ml。
25.本发明提供了一种还原催化剂的制备方法,包括以下步骤:将蛋膜粉与含有三价铁离子和二价钯离子的金属离子溶液进行第一混合,得到第一混合液;将所述第一混合液和碱进行第二混合进行氢氧化反应,得到第二混合液;将所述第二混合液和硼氢化盐溶液进行第三混合进行还原反应,得到第三混合液;将所述第三混合液固液分离,得到固体前驱物;将所述固体前驱物进行热处理,得到所述还原催化剂。本发明以蛋膜作为载体原料经过负载催化金属和热处理,制备得到负载催化活性金属的三维网状碳氮化合物,该化合物具有催化还原反应的作用,原料廉价易得,制备方法简单,降低了催化剂的制备成本。同时,由于蛋膜的结构是一种三维的纤维网状结构,能够吸附大量的金属离子,并且蛋膜中含有大量的-oh、c-o-c官能团,能够与金属离子产生强的相互作用,进而能够锚固金属,锚固金属后蛋膜经过热处理后形成保留原来网状结构的碳氮化合物,增大了具有催化活性金属和反
应物的接触面积,提高了催化效果。同时,fe元素的加入也可以起到助催化的作用,进一步提升了还原催化剂的催化效果。
26.本发明还提供了上述方案制备得到的还原催化剂。本发明提供的催化剂具有良好的催化活性,可以用于制备3-甲基-3,4-二氢-2h-1,4-苯并恶嗪。
27.本发明还提供了一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法。本发明提供的制备方法以2-丙酮氧基硝基苯为原料,还原-合环反应得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪。其中,还原-合环反应采用的催化剂为上述方案所述的还原催化剂。该制备方法操作简单,产品收率和纯度较高,适合工业化生产。
附图说明
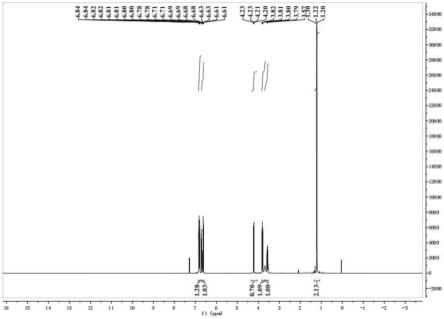
28.图1为实施例1制备得到的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的1h-nmr图。
具体实施方式
29.本发明提供了一种还原催化剂的制备方法,包括以下步骤:
30.将蛋膜粉与含有三价铁离子和二价钯离子的溶液进行第一混合,得到第一混合液;
31.将所述第一混合液和碱进行第二混合进行氢氧化反应,得到第二混合液;
32.将所述第二混合液和硼氢化盐溶液进行第三混合进行还原反应,得到第三混合液;
33.将所述第三混合液过滤,得到固体前驱物;
34.将所述固体前驱物进行热处理,得到所述还原催化剂。
35.如无特殊说明,本发明使用的制备原料均为市售。
36.本发明将蛋膜粉与含有三价铁离子和二价钯离子的溶液进行第一混合,得到第一混合液。在本发明中,所述蛋膜粉优选为鸭蛋膜粉、鹅蛋膜粉或鸡蛋膜粉,更优选为鸡蛋膜粉。在本发明中,所述蛋膜粉的制备方法优选为:将蛋壳进行第一清洗,然后将蛋壳膜从蛋壳上剥离,得到剥离后的蛋壳膜;将剥离后的蛋壳膜依次进行第二清洗、酸洗、第三清洗、干燥和研磨,得到蛋膜粉。在本发明中,所述蛋壳优选为新鲜蛋壳,所述第一清洗、第二清洗和第三清洗所用的清洗液优选为水,所述第一清洗、第二清洗和第三清洗优选采用多次清洗的方式,其中,所述第一清洗以新鲜蛋壳表面无可见蛋清和蛋黄为准,所述第二清洗以蛋壳膜表面无可见蛋清、蛋黄或蛋壳为准,所述第三清洗在每次清洗后测量清洗液的ph值,直至清洗液的ph值无变化为准。在本发明中,所述酸洗优选在室温下进行,所述酸洗用酸洗液优选为盐酸溶液,所述盐酸溶液的质量浓度优选为20~35%,所述酸洗的时间优选为12~18h。在本发明中,所述干燥的温度优选为60~70℃,所述干燥的时间优选为6~8h。本发明优选通过第一清洗和第二清洗将蛋壳膜上的蛋清和蛋黄去除,通过酸洗去除膜上残留的蛋壳,通过第三清洗以将蛋壳膜上的盐酸去除。在本发明中,所述蛋膜粉的粒径优选为20~40μm,更优选为25~35μm。
37.在本发明中,所述蛋膜粉与所述溶液的质量体积比优选为1g:(90~110)ml;所述溶液中三价铁离子的浓度优选为50~100mmol/l,更优选为60~80mmol/l,进一步优选为65~75mmol/l;所述溶液中二价钯离子的浓度为1.0~2.0mmol/l,更优选为1.2~1.8mmol/l,
进一步优选为1.4~1.6mmol/l。在本发明中,所述含有三价铁离子和二价钯离子的金属离子溶液优选由三价铁化合物、二价钯化合物和水配制得到,所述三价铁化合物优选为fecl3、fecl3·
6h2o、fe(so4)3或fe(no3)3,更优选为fecl3或fecl3·
6h2o,进一步优选为fecl3·
6h2o,所述二价钯化合物优选为na2pdcl4或h2pdcl4,,更优选为h2pdcl4,所述水优选为蒸馏水。在本发明中,所述第一混合的方式优选为搅拌,所述搅拌优选在室温下进行,所述搅拌的时间优选2~4h,更优选为3~4h。本发明将铁离子和钯离子的含量控制在上述范围内有利于使适量的铁离子和钯离子负载到蛋膜粉上,保证复合催化剂中的催化活性物质的有效负载率,保证催化剂具有较好的催化效果。
38.得到第一混合液后,本发明将所述第一混合液和碱进行第二混合进行氢氧化反应,得到第二混合液。在本发明中,所述碱优选为naoh、koh或na2co3,更优选为naoh,所述碱优选以水溶液的形式和第一混合液进行第二混合,所述水溶液的浓度优选为0.1~0.3mol/l,更优选为0.1~0.2mol/l。在本发明中,所述碱的添加量以调节第二混合液的ph值为9.5~10.5为准,所述第二混合的方式优选为搅拌,所述搅拌优选在室温下进行,所述搅拌的时间优选为1~3h,更优选为1.5~2h。本发明优选通过加碱进行第二混合生成铁和钯的氢氧化物,使其以固体的形态负载在蛋膜粉上。
39.得到第二混合液后,本发明将所述第二混合液和硼氢化盐溶液进行第三混合进行还原反应,得到第三混合液。在本发明中,所述硼氢化盐溶液优选为libh4或nabh4的水溶液,更优选为nabh4的水溶液。在本发明中,所述硼氢化盐溶液中硼氢根和三价铁离子的摩尔比优选为(0.1~0.5):1,更优选为(0.15~0.45):1,进一步优选为(0.2~0.42):1,最优选为(0.25~0.4):1。本发明优选将硼氢根的含量控制在上述范围内使混合液中铁离子和钯离子得到较为彻底的还原,有利于提高催化剂的催化性能。在本法明中,所述第三混合的方式优选为搅拌,所述搅拌优选在室温下进行,所述搅拌的时间优选为3~5h,更优选为3.5~5h,进一步优选为4~4.5h。
40.得到第三混合液后,本发明将所述第三混合液固液分离,得到固体前驱物。本发明对所述固液分离的方式没有特别要求,为本领域的常规技术手段,具体如过滤。
41.得到固体前驱物后,本发明将所述固体前驱物进行热处理,得到所述还原催化剂。在本发明中,所述热处理优选在保护气氛下进行,所述保护气氛优选为氮气气氛。在本发明中,所述热处理优选在管式炉中进行。在本发明中,所述热处理的温度优选为350~550℃,更优选为400~500℃,时间优选为1.5~4h,更优选为2~3h。在本发明中,升温至所述热处理的温度的升温速率优选为3~7℃/min,更优选为4~6℃/min,进一步优选为4~5℃/min。在本发明中,对所述固体前驱物进行热处理时一方面会使蛋壳膜碳氮化,另一方面使固体前驱物中的含铁和钯的前驱体进一步还原成铁单质与钯单质。本发明优选上述热处理的条件有利于铁单质与钯单质的生成,以提高催化剂的催化活性。
42.本发明提供了上述技术方案所述制备方法制备得到的还原催化剂。在本发明中,所述还原催化剂包括载体和负载在载体上的铁单质和钯单质,所述载体为三维网状碳氮化合物所述还原催化剂中铁单质和钯单质的负载总量优选为20~25%,更优选为22~24%,进一步优选为22.5~23%。
43.本发明还提供了一种3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的制备方法,包括如下步骤:
44.将2-丙酮氧基硝基苯、还原催化剂和溶剂混合,在氢气气氛下进行还原-合环反应,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪;
45.所述还原催化剂为上述技术方案所述还原催化剂。
46.本发明将2-丙酮氧基硝基苯、还原催化剂和溶剂混合,在氢气气氛下进行还原-合环反应,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,所述还原催化剂为上述技术方案所述还原催化剂。本发明优选先将2-丙酮氧基硝基苯、还原催化剂和溶剂在反应器中混合均匀,然后向反应器中通入氢气进行还原-合环反应。在本发明中,所述还原-合环反应的机理如式i所示:
[0047][0048]
在本发明中,所述还原催化剂的质量优选为2-丙酮氧基硝基苯质量的20~50%,更优选为20~40%,进一步优选为20~30%,最优选为20%。在本发明中,所述氢气的压力优选为0.2~0.5kpa,更优选为0.3~0.4kpa。在本发明中,所述2-丙酮氧基硝基苯与溶剂的质量体积比优选为1g:(20~30)ml,更优选为1g:(22~28)ml,进一步选为1g:(24~27)ml。在本发明中,所述溶剂优选包括甲醇、乙醇、四氢呋喃、二氯甲烷和正丙醇中的任意一种或多种,更优选包括甲醇、乙醇、四氢呋喃或二氯甲烷。
[0049]
在本发明中,所述还原-合环反应的反应温度优选为30~50℃,更优选为35~48℃,进一步优选为35~40℃,所述反应时间优选为10~20h,更优选为10~16h,进一步优选为12~15h。
[0050]
还原-合环反应完成后,本发明优选对所述还原-合环反应后的体系进行后处理,得到所述3-甲基-3,4-二氢-2h-1,4-苯并恶嗪。在本发明中,所述后处理优选包括依次进行的去除催化剂、去除溶剂和提纯。本发明优选采用过滤的方式去除催化剂,得到含3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的粗产品溶液与滤液的混合液。本发明对所述过滤的方式没有特别要求,将体系中的催化剂去除干净即可。本发明优选对所述混合液采用旋蒸的方式得到含3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的粗产品溶液。在本发明中,所述旋蒸温度优选为40~50℃,所述含3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的粗产品溶液与所述混合液的体积比优选为(0.2~0.3):1。本发明优选在硅胶柱中提纯含3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的粗产品溶液,得到高纯度的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪。在本发明中,所述硅胶柱中硅胶的粒径优选为80~100目,所述提纯所用的洗脱剂优选为石油醚和乙酸乙酯的混合溶液,所述石油醚和乙酸乙酯的体积比优选为9~11:1,更优选为10:1。
[0051]
在本发明中,所述2-丙酮氧基硝基苯为市售或自行制备,当所述2-丙酮氧基硝基苯为自行制备时,所述2-丙酮氧基硝基苯通过以下步骤制备得到:
[0052]
将邻硝基苯酚、溴丙酮、无机碱性化合物、相转移催化剂和有机溶剂混合,进行取
代反应,得到2-丙酮氧基硝基苯;
[0053]
所述取代反应的反应温度为50~70℃,反应时间为4~8h;所述相转移催化剂为季铵盐;所述无机碱性化合物包括碱金属氢氧化物、碱金属碳酸盐和碱金属碳酸氢盐的任意一种或多种;所述有机溶剂包括甲苯、丙酮、乙腈、n,n-二甲基甲酰胺和二甲亚砜中的任意一种或多种;所述邻硝基苯酚、溴丙酮和无机碱性化合物的摩尔比为1:(0.5~2):(0.5~3);所述相转移催化剂的质量为邻硝基苯酚质量的3~10%;所述邻硝基苯酚与有机溶剂的质量体积比为1g:(2~5)ml。
[0054]
在本发明中所述2-丙酮氧基硝基苯为市售为当自行制备时,所述制备3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的合成路线如式ii所示:
[0055][0056]
本发明将邻硝基苯酚、溴丙酮、无机碱性化合物、相转移催化剂和有机溶剂混合,进行取代反应,得到2-丙酮氧基硝基苯。在本发明中,所述相转移催化剂优选为季铵盐,所述季铵盐优选包括甲基三丁基氯化铵(tbmac)、苄基三乙基氯化铵(teba)、四丁基溴化铵(tbab)、四丁基氯化铵和四丁基硫酸氢铵中的任意一种或多种,更优选包括甲基三丁基氯化铵(tbmac)、苄基三乙基氯化铵或四丁基溴化铵。在本发明中,所述无机碱性化合物包括碱金属的氢氧化物、碱金属的碳酸盐和碱金属的碳酸氢盐的任意一种或多种,所述碱金属的氢氧化物优选包括氢氧化钠或氢氧化钾,更优选为氢氧化钠,所述碱金属的碳酸盐优选包括碳酸钠或碳酸钾,更优选为碳酸钾,所述碱金属的碳酸氢盐优选包括碳酸氢钠或碳酸氢钾,更优选为碳酸氢钠。在本发明中,所述有机溶剂优选包括甲苯、丙酮、乙腈、n,n-二甲基甲酰胺和二甲亚砜中的任意一种或多种,更优选包括甲苯、丙酮和乙腈中的任意一种或多种。在本发明中,所述邻硝基苯酚、溴丙酮和无机碱性化合物的摩尔比优选为1:(0.5~2):(0.5~3),更优选为1:(0.5~1.5):(0.5~2),进一步优选为1:(0.5~1):(0.5~1.1)。在本发明中,所述相转移催化剂的质量优选为邻硝基苯酚质量的3~10%,更优选为5~9%,进一步优选为6~8%。在本发明中,所述邻硝基苯酚与有机溶剂的质量体积比优选为1g:(2~5)ml,更优选为1g:(2.5~4.5)ml,进一步优选为1g:(2.6~3.5)ml。本发明优选将邻硝基苯酚、溴丙酮、无机碱性化合物和相转移催化剂溶解于有机溶剂中,具体为:先将所述邻硝基苯酚与无机碱性化合物溶解于有机溶剂中,再加入相转移催化剂和溴丙酮。
[0057]
在本发明中,所述取代反应的反应温度优选为50~70℃,更优选为55~65℃;所述取代反应的反应时间优选为4~8h,更优选为5~7h。在本发明中,在取代反应过程中,邻硝基苯酚上的r-oh在碱性条件下变为r-o-,r-o-与溴丙酮反应,得到2-丙酮氧基硝基苯。
[0058]
取代反应完成后,本发明优选对所述取代反应后的体系进行后处理,得到所述2-丙酮氧基硝基苯。在本发明中,所述后处理优选包括依次进行的淬灭、调节反应体系ph值、萃取、萃取相脱水、去除脱水剂和去除萃取剂。在本发明中,优选采用水对反应进行淬灭,所述淬灭反应用的水与反应液的体积比优选为(1~2):1,更优选为(1.2~1.8):1。本发明优选将反应体系调整为6.5~7,得到被萃取体系。在本发明中,优选采用盐酸调节体系的ph值,所述盐酸的浓度优选为10~20%。在本发明中,所述萃取用萃取剂优选为乙酸乙酯,所
述萃取剂与被萃取体系的体积比优选为(1~2):1,更优选为(1~1.5):1。所述萃取相脱水用的脱水剂优选为无水硫酸钠,所述脱水剂与所述萃取相的质量体积比优选为(0.2~0.4)g:1ml,更优选为(0.25~0.35)g:1ml。本发明优选采用过滤的方式去除脱水剂,本发明对所述过滤的方式没有特别要求,为本领域技术人员熟知的方式。本发明优选采用旋蒸去除萃取剂,得到2-丙酮氧基硝基苯固体。在本发明中,所述旋蒸的温度优选为60~70℃,更优选为64~68℃,本发明对所述旋蒸时间没有要求,将萃取剂旋蒸干净即可。
[0059]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
[0060]
实施例1
[0061]
将1g鸡蛋膜粉加入到由fecl3·
6h2o、h2pdcl4和100ml蒸馏水配制的金属离子溶液中,其中,三价铁离子的浓度为50mmol/l,二价钯离子的浓度为1.0mmol/l。在室温下搅拌2h,之后加入0.1mol/l的naoh水溶液,调整体系的ph=10,之后搅拌1h,再向体系中加入0.2mol/l的nabh4水溶液10ml,在室温下搅拌3h,搅拌后将体系过滤并洗涤,得到固体前驱物,将固体前驱物放入管式炉中在n2气氛下以5℃/min的升温速率加热到500℃保温2h,得到还原催化剂,记为pd/fe/esm催化剂,其中,钯单质和铁单质的总负载量为22%。
[0062]
向反应器中加入27.8g邻硝基苯酚、20.06g溴丙酮、18.52g碳酸氢钠、1.08g甲基三丁基氯化铵和80ml甲苯,在65℃下搅拌4h进行取代反应。反应结束后,向反应器中加入100ml水进行淬灭,之后加入浓度为15%的hcl溶液调节体系的ph值为6.5,加入100ml乙酸乙酯萃取2-丙酮氧基硝基苯,萃取完毕后,向反应器中加入20g无水硫酸钠去除水,将反应器中的混合物过滤去除干燥剂,将得到的滤液在65℃下旋蒸,得到2-丙酮氧基硝基苯,外观为为橘色固体。经计算和测定,2-丙酮氧基硝基苯的产率为92.8%,纯度为98.5%。
[0063]
向反应器中加入2g制备得到的2-丙酮氧基硝基苯、0.4g pd/fe/esm催化剂以及50ml甲醇,向反应器中通入氢气,使氢气的压力达到0.4kpa,在40℃下搅拌反应12h。反应结束后,通过过滤去除反应体系中的固体催化剂,将得到的滤液旋蒸去除大部分溶剂,得到粗产物。采用硅胶柱对粗产物进行柱层析,洗脱剂为石油醚和乙酸乙酯构成,两者体积比为10:1,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,为黄色油状液体。经计算和测定,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率为86.45%,纯度为97.3%。图1为实施例1制备得到的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的1h-nmr图。从图1可以看出,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的特征峰明显,表明采用实施例1的制备方法制备得到的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪纯度高。
[0064]
实施例2
[0065]
将2g鸡蛋膜粉加入到由fecl3·
6h2o、h2pdcl4和200ml蒸馏水配制的金属离子溶液中,其中,三价铁离子的浓度为60mmol/l,二价钯离子的浓度为1.5mmol/l。在室温下搅拌3h,之后加入0.15mol/l的naoh水溶液,调整体系的ph=10,之后搅拌2h,再向体系中加入0.3mol/l的nabh4水溶液10ml,在室温下搅拌4h,搅拌后将体系过滤并洗涤,得到固体前驱物,将固体前驱物放入管式炉中在n2气氛下以4℃/min的升温速率加热到500℃保温3h,得到还原催化剂,记为pd/fe/esm催化剂,其中,钯单质和铁单质的总负载量为21%。
[0066]
向反应器中加入14g邻硝基苯酚、10g溴丙酮、9g碳酸氢钾、0.51g苄基三乙基氯化铵和40ml丙酮,在70℃下搅拌5h进行取代反应。反应结束后,向反应器中加入50ml水进行淬灭,之后加入浓度为15%的hcl溶液调节体系的ph值为7,加入50ml乙酸乙酯萃取2-丙酮氧
基硝基苯,萃取完毕后,向反应器中加入10g无水硫酸钠去除水,将反应器中的混合物过滤去除干燥剂,将得到的滤液在70℃下旋蒸,得到2-丙酮氧基硝基苯,外观为为橘色固体。经计算和测定,2-丙酮氧基硝基苯的产率为90.0%,纯度为98.5%。
[0067]
向反应器中加入2g制备得到的2-丙酮氧基硝基苯、0.4g pd/fe/esm催化剂以及50ml乙醇,向反应器中通入氢气,使氢气的压力达到0.5kpa,在35℃下搅拌反应10h。反应结束后,通过过滤去除反应体系中的固体催化剂,将得到的滤液旋蒸去除大部分溶剂,得到粗产物。采用硅胶柱对粗产物进行柱层析,洗脱剂为石油醚和乙酸乙酯构成,两者体积比为10:1,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,为黄色油状液体。经计算和测定,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率为82%,纯度为97.1%。
[0068]
实施例3
[0069]
将2g鸡蛋膜粉加入到由fecl3·
6h2o、h2pdcl4和200ml蒸馏水配制的金属离子溶液中,其中,三价铁离子的浓度为70mmol/l,二价钯离子的浓度为1.7mmol/l。在室温下搅拌4h,之后加入0.15mol/l的naoh水溶液,调整体系的ph=10,之后搅拌1h,再向体系中加入0.3mol/l的nabh4水溶液10ml,在室温下搅拌3h,搅拌后将体系过滤并洗涤,得到固体前驱物,将固体前驱物放入管式炉中在n2气氛下以3℃/min的升温速率加热到500℃保温3h,得到还原催化剂,记为pd/fe/esm催化剂,其中,钯单质和铁单质的总负载量为20%。
[0070]
向反应器中加入26g邻硝基苯酚、20g溴丙酮、19g碳酸钠、1.1g四丁基溴化铵和80ml乙腈,在50℃下搅拌6h进行取代反应。反应结束后,向反应器中加入100ml水进行淬灭,之后加入浓度为15%的hcl溶液调节体系的ph值为6.5,加入100ml乙酸乙酯萃取2-丙酮氧基硝基苯,萃取完毕后,向反应器中加入20g无水硫酸钠去除水,将反应器中的混合物过滤去除干燥剂,将得到的滤液在65℃下旋蒸,得到2-丙酮氧基硝基苯,外观为为橘色固体。经计算和测定,2-丙酮氧基硝基苯的产率为87.3%,纯度为98.6%。
[0071]
向反应器中加入4g制备得到的2-丙酮氧基硝基苯、0.8g pd/fe/esm催化剂以及100ml四氢呋喃,向反应器中通入氢气,使氢气的压力达到0.2kpa,在50℃下搅拌反应15h。反应结束后,通过过滤去除反应体系中的固体催化剂,将得到的滤液旋蒸去除大部分溶剂,得到粗产物。采用硅胶柱对粗产物进行柱层析,洗脱剂为石油醚和乙酸乙酯构成,两者体积比为10:1,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,为黄色油状液体。经计算和测定,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率为83.8%,纯度为97.0%。
[0072]
实施例4
[0073]
将3g鸡蛋膜粉加入到由fecl3·
6h2o、h2pdcl4和300ml蒸馏水配制的金属离子溶液中,其中,三价铁离子的浓度为80mmol/l,二价钯离子的浓度为1.2mmol/l。在室温下搅拌2h,之后加入0.2mol/l的naoh水溶液,调整体系的ph=10,之后搅拌2h,再向体系中加入0.3mol/l的nabh4水溶液30ml,在室温下搅拌3h,搅拌后将体系过滤并洗涤,得到固体前驱物,将固体前驱物放入管式炉中在n2气氛下以5℃/min的升温速率加热到400℃保温3h,得到还原催化剂,记为pd/fe/esm催化剂,其中,钯单质和铁单质的总负载量为22%。
[0074]
向反应器中加入55.5g邻硝基苯酚、41g溴丙酮、36g碳酸钠、2.2g甲基三丁基氯化铵和160ml二甲亚砜,在65℃下搅拌4h进行取代反应。反应结束后,向反应器中加入100ml水进行淬灭,之后加入浓度为20%的hcl溶液调节体系的ph值为7,加入200ml乙酸乙酯萃取2-丙酮氧基硝基苯,萃取完毕后,向反应器中加入40g无水硫酸钠去除水,将反应器中的混合
物过滤去除干燥剂,将得到的滤液在65℃下旋蒸,得到2-丙酮氧基硝基苯,外观为为橘色固体。经计算和测定,2-丙酮氧基硝基苯的产率为89.5%,纯度为98.9%。
[0075]
向反应器中加入6g制备得到的2-丙酮氧基硝基苯、1.2g pd/fe/esm催化剂以及150ml二氯甲烷,向反应器中通入氢气,使氢气的压力达到0.3kpa,在50℃下搅拌反应16h。反应结束后,通过过滤去除反应体系中的固体催化剂,将得到的滤液旋蒸去除大部分溶剂,得到粗产物。采用硅胶柱对粗产物进行柱层析,洗脱剂为石油醚和乙酸乙酯构成,两者体积比为10:1,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,为黄色油状液体。经计算和测定,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率为81.2%,纯度为96.3%。
[0076]
实施例5
[0077]
将10g鸡蛋膜粉加入到由fecl3·
6h2o、h2pdcl4和1000ml蒸馏水配制的金属离子溶液中,其中,三价铁离子的浓度为90mmol/l,二价钯离子的浓度为1.4mmol/l。在室温下搅拌5h,之后加入0.1mol/l的naoh水溶液,调整体系的ph=10,之后搅拌2h,再向体系中加入0.2mol/l的nabh4水溶液100ml,在室温下搅拌4h,搅拌后将体系过滤并洗涤,得到固体前驱物,将固体前驱物放入管式炉中在n2气氛下以5℃/min的升温速率加热到500℃保温2h,得到还原催化剂,记为pd/fe/esm催化剂,其中,钯单质和铁单质的总负载量为21%。
[0078]
向反应器中加入278g邻硝基苯酚、200g溴丙酮、185g碳酸氢钠、11g甲基三丁基氯化铵和800ml甲苯,在65℃下搅拌4h进行取代反应。反应结束后,向反应器中加入1000ml水进行淬灭,之后加入浓度为20%的hcl溶液调节体系的ph值为7,加入1000ml乙酸乙酯萃取2-丙酮氧基硝基苯,萃取完毕后,向反应器中加入200g无水硫酸钠去除水,将反应器中的混合物过滤去除干燥剂,将得到的滤液在70℃下旋蒸,得到2-丙酮氧基硝基苯,外观为为橘色固体。经计算和测定,2-丙酮氧基硝基苯的产率为87.3%,纯度为98.5%。
[0079]
向反应器中加入20g制备得到的2-丙酮氧基硝基苯、4g pd/fe/esm催化剂以及500ml二氯甲烷,向反应器中通入氢气,使氢气的压力达到0.5kpa,在40℃下搅拌反应20h。反应结束后,通过过滤去除反应体系中的固体催化剂,将得到的滤液旋蒸去除大部分溶剂,得到粗产物。采用硅胶柱对粗产物进行柱层析,洗脱剂为石油醚和乙酸乙酯构成,两者体积比为10:1,得到3-甲基-3,4-二氢-2h-1,4-苯并恶嗪,为黄色油状液体。经计算和测定,3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率为82%,纯度为98%。
[0080]
对比例1
[0081]
将实施例1中的pd/fe/esm催化剂替换为铂碳,铂碳上铂的负载量为20.6%,其他条件与实施例1相同,最终得到的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率78.3%。
[0082]
对比例2
[0083]
将实施例1中的pd/fe/esm催化剂替换为钯碳,钯碳上钯的负载量为19.4%。其他条件与实施例1相同,最终得到的3-甲基-3,4-二氢-2h-1,4-苯并恶嗪的产率80.7%。
[0084]
从实施例1~5和对比例1~2的结果可以看出,本发明采用蛋膜粉制备得到的还原催化剂的催化效果优于铂碳和钯碳催化效果。这是由于蛋膜的结构是一种三维的纤维网状结构,能够吸附大量的金属离子,并且蛋膜中含有大量的-oh、c-o-c官能团,能够与金属粒子产生强的相互作用,进而能够锚固金属,锚固金属后蛋膜经过热处理后形成保留原来网状结构的碳氮化合物,增大了具有催化活性金属和反应物的接触面积,提高了催化效果。同时,fe元素的加入也可以起到助催化的作用,进一步提升了还原催化剂的催化效果。
[0085]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1