大电流电催化水分解制氢的催化剂及其制备方法和应用
1.本发明涉及催化剂的技术领域,尤其涉及多种大电流电催化水分解制氢的催化剂及其制备方法和应用。
背景技术:
2.电解水制氢是一种绿色环保、方便快捷和有巨大发展潜力的可持续制氢方式。大量研究致力于通过不同的调控策略,例如维度调控、缺陷工程、掺杂、异质结、形貌调控、利用集流体等,来降低析氢反应达到10ma
·
cm-2
的过电位及反应的塔菲尔斜率,提高阴极材料的工作稳定性,以此提升催化材料的综合性能及其可运用于工业生产的能力。然而,要想进一步满足促进电催化能力的同时提高能源利用率、降低生产成本,实现具有工业应用前景的大电流(析氢电流密度≥500ma
·
cm-2
)电催化产氢的催化剂,单一的调控策略则稍显不足。此外,相对于价格高昂、资源稀缺、电化学稳定性较差而难以实现大规模工业化应用的ru,rh,pd,ir,pt等贵金属涉及的催化剂,mn,fe,co,ni,mo,w等过渡金属催化剂,例如过渡金属硫化物、氮化物、氧化物、碳化物和磷化物等来调控电化学催化水分解产生氢气,将在大电流电催化水分解制氢领域具有更大的应用前景。
3.目前,将贵金属以高分散形式与其它金属或非金属氧化物进行联合制备具有异质结的催化剂,或直接利用过渡金属单质与同种或不同金属氧化物形成异质结界面,以此促进水在氧化物上的分解和后续氢气在贵金属或过渡金属单质位点的产生是一种常用的改性方法。该方法具有增加活性位点数目,有效促进水分解制氢,降低达到10ma
·
cm-2
电流密度的过电位等优点。但是该方法依然需要面对高昂的生产成本,或仅可实现在小电流密度下的有效产氢,稳定性差,粉末样品放大应用不方便,活性组分分散性差等问题。此外,大量研究利用泡沫镍、泡沫铜、碳纸等集流体作为电催化活性组分的载体,以此来改善催化剂的导电性、高电流强度耐受性、传质及负载量低等问题。但是该方法面临着许多优异材料无法原位生长上去,材料容易脱落,本征活性较差,催化位点较为单一等问题,从而使得析氢反应电流密度小,过电位较大,限制了其在工业条件下应用的前景。
4.因此,如何制备具有高分散、高活性、低电位且能同时满足小电流密度和大电流密度应用条件的电催化水分解催化剂是非常重要的。
技术实现要素:
5.针对现有技术中存在的上述问题,本发明公开了多种大电流电催化水分解制氢的催化剂,该催化剂具有多种活性组分,且多种活性组分间存在协同效应,各组分间紧密结合形成了多极串联的结构,从而暴露出大量的催化活性位点,提高导电能力以及具有高强度的稳固性。作为应用,该催化剂在大电流密度下电催化产氢反应中具有优异的催化性能和长程稳定性。
6.具体技术方案如下:
7.本发明公开了三种大电流电催化水分解制氢催化剂,活性组分的组成分别为co/
coo/comoo3、coo/comoo4和como/coo/comoo3。
8.第一种催化剂,co/coo/comoo3,包括集流体,和原位生长于所述集流体上的活性组分;
9.所述活性组分包括呈棒状的comoo3,以及均匀生长在棒状的comoo3上的co纳米颗粒和coo纳米颗粒。
10.优选的:
11.所述棒状的comoo3的长度为11~30μm,横截面为0.92~2.82μm2;
12.co纳米颗粒和coo纳米颗粒的平均粒径独立的选自10~50nm。
13.以所述催化剂的质量计,所述活性组分的质量分数为50~70%;
14.所述集流体选自泡沫镍。
15.第二种催化剂,coo/comoo4,包括集流体,和原位生长于所述集流体上的活性组分;
16.所述活性组分包括呈棒状的comoo4,以及均匀生长在棒状的comoo4上的coo纳米颗粒。
17.优选的:
18.所述棒状的comoo4的长度为11~30μm,横截面为0.92~2.82μm2;
19.所述coo纳米颗粒的平均粒径为10~50nm。
20.以所述催化剂的质量计,所述活性组分的质量分数为50~70%;
21.所述集流体选自泡沫镍。
22.第三种催化剂,como/coo/comoo3,包括集流体,和原位生长于所述集流体上的活性组分;
23.所述活性组分包括呈棒状的comoo3,以及均匀生长在棒状的comoo3上的como纳米颗粒和coo纳米颗粒。
24.优选的:
25.所述棒状的comoo3的长度为11~30μm,横截面为0.92~2.82μm2;
26.como纳米颗粒与coo纳米颗粒的平均粒径独立地选自10~50nm。
27.以所述催化剂的质量计,所述活性组分的质量分数为50~70%;
28.所述集流体选自泡沫镍。
29.本发明还公开了上述三种催化剂的制备方法,包括:
30.(1)将金属钴盐、钼酸铵与水混合,向混合液中加入集流体,经水热反应得到金属前驱体;
31.所述水热反应的温度为140~160℃,时间为1~10h;
32.(2)将步骤(1)制备的金属前驱体在惰性气氛或还原气氛下进行高温焙烧处理,得到所述大电流电催化水分解制氢催化剂;
33.所述高温焙烧处理的温度为200~600℃,时间为20min~8h。
34.步骤(1)中:
35.所述金属钴盐选自钴的硝酸盐、氯化物、乙酰丙酮盐、偏钒酸盐中的一种或多种;
36.所述混合液中,钼酸铵的浓度为0.001~0.1mol/l;浓度过低,无法在集流体上充分生长致密的钼酸盐前驱体;浓度过高,生长于集流体上的钼酸盐前驱体过于厚重,在后续
处理及反应中易塌陷,造成结构的破坏,且原料利用率低。
37.金属钴盐与钼酸铵的摩尔比为1~6:1;金属含量过少,则活性位点少,催化效率低;金属含量过多,则无法有效利用金属,且分散较差。
38.水热反应后还需进行洗涤与干燥的后处理。
39.步骤(1)中:
40.所述集流体在使用之前优选进行预处理,具体包括:
41.通过酸和醇的混合液对集流体进行超声预处理,去除表面的氧化物层及有机物;再将集流体浸入去离子水中进一步超声处理去除残留的酸和醇混合物,处理后的集流体浸入去离子水中,备用。
42.优选的,所述预处理的时间为30分钟酸醇超声清洗及30分钟去离子水水超声清洗。此时长下有利于充分去除集流体表面的氧化物和有机物,以及第一步酸醇处理残留的酸与醇,又能避免长时间酸洗破坏集流体的结构。
43.进一步优选,采用质量分数为36.0~38.0%的浓盐酸与无水乙醇进行混合,酸醇体积比为1:2。
44.优选的,所述集流体选自泡沫镍。经试验发现,若将泡沫镍替换为与其性能类似的金属类集流体,如泡沫铜,按照本发明公开的制备工艺原位制备的催化剂,在大电流下析氢性能较差,但在小电流下仍具有一定的析氢性能。
45.步骤(1)中:
46.反应温度与保温时间对金属前驱体的形貌、尺寸和分布具有较大影响。优选的,所述水热反应的时间为4~6h;更优选,水热反应的温度为150℃。在上述水热条件下制备得到的金属前驱体在集流体上生长致密,呈现棒状形貌。
47.步骤(2)中:
48.优选的:
49.所述惰性气氛为ar;
50.所述还原气氛选自h2或h2/ar的混合气。
51.当采用惰性气氛,气流速度为100~500ml/min,高温焙烧处理的升温速率为5~10℃/min。
52.当采用还原气氛,气流速度为100~500ml/min,高温焙烧处理的升温速率为1~5℃/min。
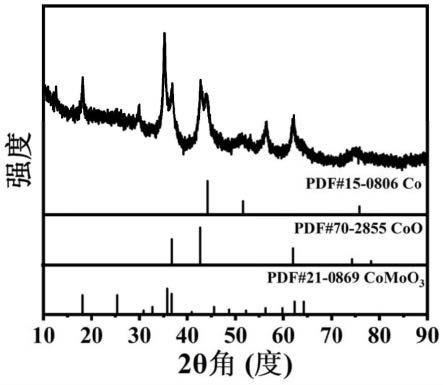
53.本发明步骤(2)的高温焙烧过程为制备不同组成催化剂的关键步骤,不同的焙烧温度所得到的催化剂的组分不同,催化性能差异较大。
54.所述高温焙烧处理的温度为200~600℃,焙烧温度过低,材料还原程度低,活性组分无法完全还原出来;焙烧温度过高,材料还原程度过高,导致其它非活性金属元素还原出来占据活性位点;保温时间为20min~8h,保温时间过短,材料还原程度低,活性组分不能充分还原出来;保温时间过长,活性组分堆积,分散度差,影响材料性能;优选的,保温时间为1~4h。
55.而当高温焙烧温度为200~400℃时,制备得到的催化剂中的活性组分为coo/comoo4;优选的,高温焙烧温度为400℃,保温时间为2h。
56.当高温焙烧温度为500℃时,制备得到的催化剂中的活性组分为co/coo/comoo3,
优选的,保温时间为2h。
57.当高温焙烧温度为600℃时,制备得到的催化剂中的活性组分为como/coo/comoo3,优选的,保温时间为2h。
58.进一步优选,所述高温焙烧温度为500℃,此时制备得到的催化剂中的活性组分为co/coo/comoo3。经试验发现,该活性组分的催化剂具有更为优异的大电流电催化水分解制氢催化活性,其在中性(ph=7)和碱性(ph=14)条件下均能达到1.3a
·
cm-2
的电流密度,远超过工业级别所需的500ma
·
cm-2
。
59.本发明还公开了上述的催化剂在大电流电催化水分解制氢反应中的应用,所述大电流电催化水分解制氢反应中,析氢电流密度≥500ma
·
cm-2
。
60.事实上,所述三种催化剂在小电流与大电流催化水解制氢反应中均具有优异的催化活性,以及优异的大电流长程稳定性,可以满足工业化电解水产氢的要求。
61.与现有技术相比,本发明具有如下优点:
62.(1)本发明采用简单且易于放大的水热法制备得到原位生长在集流体上的金属前驱体,后经过高温煅烧即可得到最终材料,操作简单,过程可控,便于工业上扩大化生产。
63.(2)本发明制备得到的催化剂具有多极串联结构,包括纳米颗粒状的活性组分高度分散在呈棒状的活性组分上,再整体原位生长并负载于集流体上,在增加导电性、增强材料的整体强度的同时也提高了催化效率。
64.(3)本发明制备得到的催化剂中,不同组成的纳米颗粒活性组分间存在协同作用,并均匀分散于呈棒状的活性组分上,共同促进水的解离与氢气的发生和析出,有效提高催化活性。
65.(4)本发明制备的具有多极串联结构的催化剂作为三电极体系的阴极,分别在碱性条件(1mol/l的koh碱性溶液)和中性条件(1mol/l的pbs中性缓冲溶液)中均具有优异的电化学催化水分解产氢性能和稳定性,尤其是在大电流密度下具有突出的电解水性能及稳定性。
附图说明
66.图1为实施例1制备的co/coo/comoo3的xrd图;
67.图2为实施例1制备的co/coo/comoo3的sem图(a~c)和hrtem图(d);
68.图3为实施例2制备的coo/comoo4的xrd图;
69.图4为实施例2制备的coo/comoo4的sem图(a~c)和hrtem图(d);
70.图5为实施例3制备的como/coo/comoo4的xrd图;
71.图6为实施例3制备的como/coo/comoo4的sem图(a~c)和hrtem图(d~f);
72.图7为实施例1~3分别制备的催化剂在碱性(ph=14)与中性(ph=7)条件下的her极化曲线图;
73.图8为实施例1~3分别制备的催化剂在碱性(ph=14)与中性(ph=7)条件下的her塔菲尔图;
74.图9为实施例1制备的co/coo/comoo3在析氢反应中的稳定性测试;
75.图10为实施例1制备的co/coo/comoo3在稳定性测试后的sem图;
76.图11为实施例4制备的co/coo/comoo
3-20min的xrd图;
77.图12为实施例4制备的co/coo/comoo
3-20min的sem图;
78.图13为实施例5制备的co/coo/comoo
3-8h的xrd图;
79.图14为实施例5制备的co/coo/comoo
3-8h的sem图;
80.图15为实施例1、4~5分别制备的催化剂在中性(ph=7)与碱性(ph=14)条件下的her极化曲线图;
81.图16为实施例1、4~5分别制备的催化剂在中性(ph=7)与碱性(ph=14)条件下的her塔菲尔图;
82.图17为对比例1制备的ceni3/ceo2/moo
x
的xrd图;
83.图18为对比例1制备的ceni3/ceo2/moo
x
的sem图;
84.图19为对比例2制备的cu/moo2的xrd图;
85.图20为对比例2制备的cu/moo2的sem图;
86.图21为对比例3制备的y2o3/ymoo4的xrd图;
87.图22为对比例3制备的y2o3/ymoo4的sem图;
88.图23为对比例4制备的zno/znmoo3/zn2mo3o8的xrd图;
89.图24为对比例4制备的zno/znmoo3/zn2mo3o8的sem图;
90.图25为实施例1与对比例1~4分别制备的催化剂在中性(ph=7)与碱性(ph=14)条件下的her极化曲线图;
91.图26为实施例1与对比例1~4分别制备的催化剂在中性(ph=7)与碱性(ph=14)条件下的her塔菲尔图。
具体实施方式
92.为了进一步理解本发明,以下结合附图及实施例对本发明提供的一种大电流电催化水分解制氢的催化剂及其制备方法和应用进行具体描述,但本发明并不限于这些实施例。该领域熟练技术人员根据上述发明内容对核心思想指导下所作的非本质的改变,仍然属于本发明的保护范围。
93.实施例1
94.将15ml质量分数为36.0%的浓盐酸与30ml无水乙醇混合,并搅拌均匀,后将一块2cm
×
4cm
×
1.5mm的泡沫镍集流体浸入其中,超声处理30min,除去表面的金属氧化物及有机物。取出泡沫镍,用去离子水洗涤三次,并置于45ml去离子水中超声30min,除去残留的盐酸与乙醇,备用。
95.将0.6mmol四水合钼酸铵和2.4mmol六水合硝酸钴溶于60ml的去离子水中,充分搅拌使其溶解。将混合液转移到100ml反应釜中,加入经上述预处理后的泡沫镍,加热至150℃反应6h。反应结束后自然冷却至室温,取出块体材料并用去离子水洗涤三次,置于70℃烘箱中干燥1h得到金属前驱体块体材料。
96.将金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气(氢氩体积比1/9)还原气氛下,以5℃/min的升温速率加热至500℃,保温2h,后自然冷却至室温,得到活性组分高分散且具有多级串联结构的大电流电催化水分解制氢的催化剂,记为co/coo/comoo3。经计算,以催化剂的总重量计,本实施制备的催化剂中活性组分的总负载量为62.65wt%。
和实施例3制备的como/coo/comoo3在相同条件下的过电位和塔菲尔斜率(实施例2:ph=7,45mv,53mv/dec;ph=14,23mv,45mv/dec;实施例3:ph=7,33mv,41mv/dec;ph=14,21mv,41mv/dec),在低电流密度下也表现出了优异的析氢性能。此外,实施例1制备的co/coo/comoo3在大电流密度下的表现更为突出,在ph=7或ph=14的条件下均能达到1.3a
·
cm-2
的电流密度,远超过工业级别所需的500ma
·
cm-2
,且所需过电位仅为379mv或173mv。在500ma
·
cm-2
下,在ph=7或ph=14的条件下,所需过电位仅为209mv或95mv。
110.相同条件下(500ma
·
cm-2
),实施例2制备的coo/comoo4和实施例3制备的como/coo/comoo3也能够达到工业级别的应用条件(实施例2:ph=7,377mv;ph=14,189mv;实施例3:ph=7,227mv;ph=14,175mv),展现出了非常好的工业应用前景。
111.图9是实施例1制备的co/coo/comoo3在碱性(ph=14)条件下,固定电位-1.18v,析氢电流密度约为500ma
·
cm-2
的稳定性测试。计时电流图表明co/coo/comoo3在固定电位-1.18v,即析氢过电位107mv下所达到的电流密度几乎保持不变,且能够保持400h以上,表明co/coo/comoo3具有优异的大电流长程稳定性,满足工业化电解水产氢的要求。
112.图10为实施例1制备的co/coo/comoo3在稳定性测试后的sem图。可以看出co/coo/comoo3在大电流密度下反应400h后依然保持较好的原始形貌特征,进一步表明co/coo/comoo3具有优异的稳定性。
113.实施例4
114.采用实施例1中制备的金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气(氢氩体积比1/9)还原气氛下,以5℃/min的升温速率加热至500℃,保温20min,后自然冷却至室温,得到的催化剂记为co/coo/comoo
3-20min。
115.图11为本实施例制备催化剂的xrd图。可以看到,本实施例制备的催化剂由co、coo及comoo3三种物相组成。
116.图12为本实施例制备催化剂的sem图。可以看出金属前驱体在500℃条件下煅烧可以得到与实施例1中类似的产物形貌,但因保温时间短,棒状活性组分载体comoo3上的颗粒小且数量少,活性位则相对较少。经统计,在与实施例1中相同面积范围内,该材料表面颗粒的平均数目为62,平均粒径为13.2nm。
117.实施例5
118.采用实施例1中制备的金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气(氢氩体积比1/9)还原气氛下,以5℃/min的升温速率加热至500℃,保温8h,后自然冷却至室温,得到的催化剂记为co/coo/comoo
3-8h。
119.图13为本实施例制备催化剂的xrd图。可以看到,本实施例制备的催化剂由co、coo及comoo3三种物相组成。
120.图14为本实施例制备催化剂的sem图。可以看出500℃下保温8h所得材料表面的颗粒急剧增大,颗粒间出现聚集现象,分散度较差。这说明活性颗粒的分散度受保温时间的影响较大。经统计,在与实施例1中相同面积范围内,该材料表面颗粒的平均数目为90,平均粒径为26.6nm。
121.应用例2
122.将实施例4制备的co/coo/comoo
3-20min和实施例5制备的co/coo/comoo
3-8h按照应用例1中的操作要求进行析氢性能的测试。
123.如图15、16所示,实验结果表明,实施例4的co/coo/comoo
3-20min和实施例5的co/coo/comoo
3-8h无论在小电流密度10ma
·
cm-2
下(实施例4:ph=7,29mv;ph=14,17mv;实施例5:ph=7,46mv;ph=14,18mv)还是大电流密度下500ma cm-2
(实施例4:ph=7,270mv;ph=14,180mv;实施例5:ph=7,419mv;ph=14,152mv),仍具有优异的析氢性能,能达到工业的应用条件。但相比于实施例1制备的co/coo/comoo3,活性颗粒较少的实施例4的co/coo/comoo
3-20min和分散度较差的实施例5的co/coo/comoo
3-8h析氢性能均有所下降,塔菲尔斜率有所增大(实施例4:ph=7,43mv/dec;ph=14,28mv/dec;实施例5:ph=7,62mv/dec;ph=14,30mv/dec),表明颗粒数目与大小和颗粒分散度均会影响析氢性能。
124.对比例1
125.将0.6mmol四水合钼酸铵和2.4mmol六水合硝酸铈溶于60ml的去离子水中,充分搅拌使其溶解。将混合液转移到100ml反应釜中,加入一块经预处理的2cm
×
4cm
×
1.5mm的泡沫镍(预处理工艺与实施例1中相同),加热至150℃反应6h。反应结束后自然冷却至室温,取出块体材料并用去离子水洗涤三次,置于70℃烘箱中干燥1h得到金属前驱体块体材料;将金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气(氢氩体积比1/9)还原气氛下,以5℃/min的升温速率加热至500℃,保温2h,后自然冷却至室温,得到水分解制氢的催化剂记为ceni3/ceo2/moo
x
。
126.图17为本对比例制备催化剂的xrd图。可以看到,本对比例制备的催化剂由ceni3、ceo2及无定形的moo
x
三种物相组成。
127.图18为本对比例制备催化剂的sem图。可以看出,本对比例制备的催化剂的主体形貌为花状,且花瓣上有少许颗粒,但自生长的花状材料在泡沫镍上的覆盖度较差。
128.对比例2
129.将0.6mmol四水合钼酸铵和2.4mmol硝酸铜水合物溶于60ml的去离子水中,搅拌溶解后转移到100ml反应釜中,加入一块经预处理的2cm
×
4cm
×
1.5mm的泡沫镍(预处理工艺与实施例1中相同),加热至150℃反应6h。反应结束后自然冷却至室温,取出块体材料洗涤并干燥1h得到金属前驱体块体材料;将金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气中以5℃/min的升温速率加热至500℃,保温2h,后自然冷却至室温,得到水分解制氢的催化剂记为cu/moo2。
130.图19为本对比例制备催化剂的xrd图。可以看到,本对比例制备的催化剂由cu、moo2两种物相组成。
131.图20为本对比例制备催化剂的sem图。可以看出,本对比例制备的催化剂的主体形貌为大小不均匀的球体,且球体由颗粒堆积成的刺状构成。
132.对比例3
133.将0.6mmol四水合钼酸铵和2.4mmol六水合硝酸钇溶于60ml的去离子水中,搅拌溶解后转移到100ml反应釜中,加入一块经预处理的2cm
×
4cm
×
1.5mm的泡沫镍(预处理工艺与实施例1中相同),加热至150℃反应6h。反应结束后自然冷却至室温,取出块体材料洗涤并干燥1h得到金属前驱体块体材料;将金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气中以5℃/min的升温速率加热至500℃,保温2h,后自然冷却至室温,得到水分解制氢的催化剂记为y2o3/ymoo4。
134.图21为本对比例制备催化剂的xrd图。可以看到,本对比例制备的催化剂由y2o3、
ymoo4两种物相组成。
135.图22为本对比例制备催化剂的sem图。可以看到,本对比例制备的催化剂的主体形貌为相互交联的片层结构,且片层表面具有分散均匀的颗粒。
136.对比例4
137.将0.6mmol四水合钼酸铵和2.4mmol六水合硝酸锌溶于60ml的去离子水中,搅拌溶解后转移到100ml反应釜中,加入一块经预处理的2cm
×
4cm
×
1.5mm的泡沫镍(预处理工艺与实施例1中相同),加热至150℃反应6h。反应结束后自然冷却至室温,取出块体材料洗涤并干燥1h得到金属前驱体块体材料;将金属前驱体块体材料置于真空管式炉中,在300ml/min的h2/ar混合气中以5℃/min的升温速率加热至500℃,保温2h,后自然冷却至室温,得到水分解制氢的催化剂记为zno/znmoo3/zn2mo3o8。
138.图23为本对比例制备催化剂的xrd图。可以看到,本对比例制备的催化剂由zno、znmoo3和zn2mo3o8三种物相组成。
139.图24为本对比例制备催化剂的sem图。可以看到,本对比例制备的催化剂由棒状znmoo3/zn2mo3o8及其表面的zno颗粒组成。
140.对比例5
141.制备工艺与实施例1中完全相同,区别仅在于将泡沫镍集流体替换为相同尺寸的泡沫铜。
142.经xrd表征,制备得到的催化剂由co、coo及comoo3三种物相组成。
143.应用例3
144.将对比例1制备的ceni3/ceo2/moo
x
(简写为moce)、对比例2制备的cu/moo2(简写为mocu)、对比例3制备的y2o3/ymoo4(简写为moy)、对比例4制备的zno/znmoo3/zn2mo3o8(简写为mozn)和实施1制备的co/coo/comoo3(简写为moco)按照应用例1中的操作要求进行析氢性能的测试。
145.如图25、26所示,实验结果表明,对比例1~4制备的催化剂在中性条件下的析氢反应电流密度达到10ma
·
cm-2
所需的过电位和塔菲尔斜率分别为60mv、83mv、53mv、37mv和98mv/dec、104mv/dec、76mv/dec、67mv/dec,远大于实施例1制备的co/coo/comoo3的29mv和39mv/dec,表明在中性条件下,对比例1~4制备的催化剂相较于实施例1制备的co/coo/comoo3具有较差的析氢性能,且无法实现工业级别下500ma
·
cm-2
的电流密度。在碱性条件下,对比例1~4制备的催化剂实现电流密度10ma
·
cm-2
时所需的过电位和塔菲尔斜率分别为53mv、15mv、16mv、9mv和94mv/dec、41mv/dec、49mv/dec、44mv/dec,从中可以看出,对比例1制备的mocu在碱性且低电流密度下的析氢性能远低于实施例1的9mv,对比例2~4制备的催化剂的性能接近但仍低于或等于实施例1的性能。但随着电流密度的增大,对比例1~4分别制备的催化剂的析氢过电位增加比实施例1的更加快速,表明其大电流密度析氢性能逐渐不占优势,且无法同时实现在中性和碱性条件下实现工业化应用,应用范围较窄。
146.将对比例5制备的催化剂按照应用例1中的操作要求进行析氢性能的测试,实验结果表明,因活性组分在泡沫铜上的覆盖度较低,结合不够紧密,其在小电流密度10ma
·
cm-2
下的过电位(ph=7,100mv)和塔菲尔斜率(ph=7,126mv/dec)相较于实施例1均有所上升,但在同类非贵金属催化剂中表现出了较好的中性析氢性能。
147.此外应理解,在阅读了本发明的上述描述内容之后,本领域技术人员可以对本发
明作各种改动或者修改,这等价形式同样弱于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1