一种Ni-VC单原子合金电催化CO2还原材料及其制备方法
一种ni-vc单原子合金电催化co2还原材料及其制备方法
技术领域
1.本发明涉及一种ni-vc单原子合金电催化co2还原材料及其制备方法,属于复合材料制备技术领域。
背景技术:
2.目前为了抑制气候条件恶化,都鼓励发展用于减少大气中二氧化碳的排放的新技术,限制/消除二氧化碳的过度排放对全球气候的负面影响。在众多减少co2含量的途径中,co2电还原反应(co2rr)因其温和的反应条件以及较高的能源效率收到广泛关注及研究。一氧化碳(co)作为一种具有较高商业价值的co2rr还原产物,是生产碳氢化学品的重要原料,亦是众多co2rr产物中最接近商业应用的化学品。
3.镍(ni)基催化剂因其制备成本较低、储量丰富的特点被广泛用于还原产物co的研究。目前已报道的ni基催化剂均虽表现出一定的催化活性,但其ni活性位点的利用效率仍亟待提高。与此同时,如何简便高效地实现ni活性位点的电子结构优化调节仍需进一步深入研究。因此,探究一种简便高效制备具有优异co活性及选择性的ni基co2还原材料具有十分重要的实际应用意义。
技术实现要素:
4.技术问题:
5.本发明提供一种高效催化co2还原为co的ni基催化剂,避免反应过程中竞争产物h2过多的问题并提高其活性位点利用率,选取合适的方法来优化其催化性能。
6.技术方案:
7.为解决上述问题,本发明提供了一种基于静电纺丝纳米纤维为纳米反应器,利用高温热驱动作用制备得到一种ni元素原子级分散于vc(碳化钒)纳米颗粒的单原子合金(ni-vc saa)催化剂,以用于高效电催化co2还原材料。利用简便高效的高温热驱动作用将纳米纤维中的ni元素由纳米级分散转为原子级分散于vc纳米颗粒表面,制得ni-vc单原子合金电催化co2还原材料。该催化剂廉价易制,所得ni-vc saa电催化co2还原材料在较宽的电压测试范围内(-0.78~-1.18v vs.rhe),其产物co的法拉第效率能够达到近100%。
8.本发明的第一个目的是提供一种制备ni-vc saa电催化co2还原材料的方法,所述制备方法包括如下步骤:
9.(1)将镍盐、钒盐和纳米纤维前驱体均匀分散于有机溶剂中,制得静电纺丝溶液;随后利用静电纺丝制得含有镍盐与钒盐的纳米纤维膜;
10.(2)将步骤(1)所制得纳米纤维膜进行预氧化处理,随后在惰性气体氛围下进行高温碳化处理,制得ni-vc/cnfs电催化材料。
11.在本发明的一种实施方式中,步骤(1)所述的纳米纤维前驱体为聚乙烯吡咯烷酮、聚乙烯醇、聚丙烯腈中的至少一种。
12.在本发明的一种实施方式中,步骤(1)所述的镍盐为氯化镍、硝酸镍、乙酸镍、乙酰
丙酮镍中的至少一种。
13.在本发明的一种实施方式中,步骤(1)所述的钒盐为乙酰丙酮钒、氯化钒、乙酰丙酮氧钒中的至少一种。
14.在本发明的一种实施方式中,步骤(1)所述的有机溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、乙醇、水中的至少一种。
15.在本发明的一种实施方式中,步骤(1)所述的静电纺丝溶液中镍盐与钒盐的摩尔比为(0.5~3):1。优选1:1。
16.在本发明的一种实施方式中,步骤(1)所述的静电纺丝溶液中金属盐的摩尔浓度为20-50mol%。
17.在本发明的一种实施方式中,步骤(1)所述的静电纺丝溶液中金属盐的质量浓度为15~30%。
18.在本发明的一种实施方式中,步骤(1)所述的静电纺丝的条件为:纺丝正电压为10~25kv,接收器与针头的距离为10~25cm,溶液流速为0.10~0.25ml/min。
19.在本发明的一种实施方式中,步骤(2)所述的预氧化过程中,升温速率为2~30℃/min,恒温温度为200~280℃,恒温时间为0.5~3h。
20.在本发明的一种实施方式中,步骤(2)所述的高温碳化处理的条件为:升温速率为2~30℃/min,恒温温度为800~1200℃,恒温时间为0.5~3h。
21.在本发明的一种实施方式中,高温碳化处理的温度优选为1100℃。
22.在本发明的一种实施方式中,所述制备方法具体包括如下步骤:
23.(1)将硝酸镍、乙酰丙酮钒和聚乙烯吡咯烷酮加入到n,n-二甲基甲酰胺中,磁力搅拌均匀后,利用静电纺丝技术将该溶液进行纺丝,制得含有镍盐与钒盐的纳米纤维膜;
24.(2)将步骤(1)制得的含有镍盐与钒盐的纳米纤维膜进行热处理,在空气氛围下,先以2~30℃/min的升温速率升温至200~280℃,随后恒温0.5~3h进行预氧化处理;预氧化结束后,将气体氛围改为惰性气体,继续利用管式炉以5~30℃/min的升温速率升温至800~1200℃,并恒温0.5~3h进行高温碳化处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,即制得ni-vc saa电催化co2还原材料。
25.在本发明的一种实施方式中,所述步骤(2)中高温碳化处理为将纳米纤维膜夹于石墨片中,并将其置于管式炉的中间位置进行处理。
26.本发明的第二个目的是利用上述方法提供一种ni-vc saa电催化co2还原材料。
27.在本发明的一种实施方式中,所述镍元素在电催化co2还原材料中以单原子形式存在。
28.在本发明的一种实施方式中,所述钒元素在电催化co2还原材料中以vc纳米颗粒形式存在。
29.本发明的第三个目的是将上述电催化co2还原材料应用于催化co2生产一氧化碳产物中。
30.本发明的第四个目的是提供一种电催化co2还原生产一氧化碳产物的方法,所述方法是利用上述的电催化co2花园材料作为催化剂。
31.在本发明的一种实施方式中,所述电催化co2还原材料在较宽的电压测试范围内(-0.78~-1.18v vs.rhe),其产物一氧化碳的法拉第效率近100%。
32.有益效果:
33.本发明利用热驱动作用使得镍元素原子级锚定于碳化钒纳米颗粒表面,简便高效地制得ni-vc saa电催化co2还原材料,该方法简易且具有可重复性。
34.本发明所制备地ni-vc saa电催化co2还原材料因具有独特的单原子合金结构而使其具有十分优异的催化性能,其还原产物一氧化碳的法拉第效率近100%,产物选择性十分出色。
附图说明
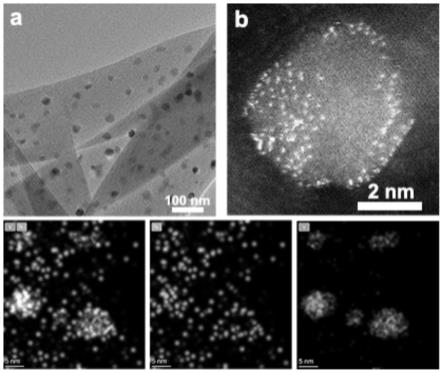
35.图1为ni-vc saa电催化co2还原材料的微观形貌图;其中(a)为ni-vc saa电催化co2还原材料的透射电镜图;(b)ni-vc saa电催化co2还原材料的球差电镜图及元素分布图。
36.图2为ni-vc saa电催化co2还原材料的x射线衍射图。
37.图3为ni-vc saa电催化co2还原材料的产物法拉第效率图。
38.图4为ni-vc saa-800电催化co2还原材料的产物法拉第效率图。
39.图5为ni
1-vc
2 saa电催化co2还原材料的产物法拉第效率图。
40.图6为ni sa电催化co2还原材料的产物法拉第效率图。
41.图7为vc电催化co2还原材料的产物法拉第效率图。
42.图8为ni sa-vc电催化co2还原材料的产物法拉第效率图。
具体实施方式
43.为了更好地理解本发明,下面结合实例进一步阐明本发明的内容,但本发明的内容不局限于下面所列实例。
44.实施例1:
45.(1)取0.4mmol氯化镍、0.4mmol乙酰丙酮钒和4g聚乙烯吡咯烷酮加入到20ml n,n-二甲基甲酰胺溶液中,在室温条件下,通过磁力搅拌12h制得均一透明的纺丝溶液,然后利用静电纺丝技术对该溶液进行纺丝,设置纺丝正极电压为18kv,接收器于纺丝针头间距为20cm,溶液流速为0.2ml/min,制得含有镍盐与钒盐的纳米纤维膜。
46.(2)将上述纳米纤维膜裁剪为5cm
×
2cm的矩形,随后将其夹于石墨片中,置于管式炉的中间部位。在空气氛围下,先以10℃/min的升温速率升温至220℃,随后恒温1h进行预氧化处理;预氧化结束后,将气体氛围改为氩气氛围,流速设置为100ml/min,继续利用管式炉以10℃/min的升温速率升温至1100℃,并恒温2h进行高温热驱动处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,即制得ni-vc saa电催化co2还原材料。
47.利用透射电镜与球差电镜对ni-vc saa催化剂的微观形貌进行研究。图1(a)为ni-vc saa的透射电镜图,由图1(a)可以观察到ni-vc saa纳米晶均匀地分布在呈三维网状的碳纳米纤维表面,纳米晶尺寸直径约为10~20nm,碳纤维的直径约为100~200nm。图1(b)为ni-vc saa的高角度环形暗场-扫描透射电镜图和元素分布图,由图1(b)可以观察到原子级分布的ni单原子锚定于vc纳米晶表面,此外元素分布图进一步表明ni的原子级分布状态。
48.对制得的ni-vc saa电催化co2还原材料进行x射线衍射表征,图2为ni-vc saa的x射线衍射图,由图2可以看出,ni-vc saa仅在37.8
°
、43.6
°
、63.2
°
、75.6
°
以及79.4
°
处产生衍射峰,上述衍射峰对应fcc相的vc纳米晶。并未观察到ni元素的相关衍射峰,表明ni元素
的原子级分布。
49.电催化二氧化碳还原性能测试在室温条件下利用电化学工作站进行分析。将制得的ni-vc/cnfs裁剪为0.8
×
0.8cm2的规则方形,因其具有自支撑性可直接用作工作电极,利用h型电解池,ag/agcl电极为参比电极,pt丝作为对电极,三者组成标准三电极体系。使用0.1m khco3作为电解液,测试过程中持续通入纯度为99.999%的co2气体,气体流速设置为20ml/min。利用电化学工作站,测试ni-vc saa电催化co2还原材料的催化活性。
50.图3为ni-vc saa电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所述催化剂在不同电压下的产物法拉第效率数值,如图3所示。在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率均超过98%,当电压为-0.98v时,产物一氧化碳的法拉第效率达到最优值,为99.6%。即使电压升高(≥-1.08v),所述催化剂仍能够有效地抑制析氢竞争反应,产物一氧化碳法拉第效率仍超过98%。总体来看,所述催化剂表现出十分优异的二氧化碳还原活性及一氧化碳选择性。
51.实施例2:
52.相比实施例1,高温热驱动温度不同:
53.取0.4mmol氯化镍、0.4mmol乙酰丙酮钒和4g聚乙烯吡咯烷酮加入到20ml n,n-二甲基甲酰胺溶液中,在室温条件下,通过磁力搅拌12h制得均一透明的纺丝溶液,然后利用静电纺丝技术对该溶液进行纺丝,设置纺丝正极电压为18kv,接收器于纺丝针头间距为20cm,溶液流速为0.2ml/min,制得含有镍盐与钒盐的纳米纤维膜。
54.将上述纳米纤维膜裁剪为5cm
×
2cm的矩形,随后将其夹于石墨片中,置于管式炉的中间部位。在空气氛围下,先以10℃/min的升温速率升温至220℃,随后恒温1h进行预氧化处理;预氧化结束后,将气体氛围改为氩气氛围,流速设置为100ml/min,继续利用管式炉以10℃/min的升温速率升温至800℃,并恒温2h进行高温热驱动处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,即制得ni-vc saa电催化co2还原材料。
55.按照实施例1中的方法测试ni-vc saa电催化co2还原材料的催化性能。
56.图4为ni-vc saa电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所述催化剂在不同电压下的产物法拉第效率数值,如图4所示。在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率为52.6~76.8%,当电压为-0.98v时,产物一氧化碳的法拉第效率达到最优值,为76.8%。电压升高(-1.18v),所述催化剂不能够高效地抑制析氢竞争反应,产物一氧化碳法拉第效率为52.6%。
57.同样的,参照实施例1,仅改变高温热驱动温度,分别替换为900、1000、1200、1500℃,相应制得的电催化材料的催化性能如表1所示。
58.表1
[0059][0060]
[0061]
实施例3:
[0062]
相比实施例1,金属盐用量不同:
[0063]
取0.2mmol氯化镍、0.4mmol乙酰丙酮钒和4g聚乙烯吡咯烷酮加入到20ml n,n-二甲基甲酰胺溶液中,在室温条件下,通过磁力搅拌12h制得均一透明的纺丝溶液,然后利用静电纺丝技术对该溶液进行纺丝,设置纺丝正极电压为18kv,接收器于纺丝针头间距为20cm,溶液流速为0.2ml/min,制得含有镍盐与钒盐的纳米纤维膜。
[0064]
将上述纳米纤维膜裁剪为5cm
×
2cm的矩形,随后将其夹于石墨片中,置于管式炉的中间部位。在空气氛围下,先以10℃/min的升温速率升温至220℃,随后恒温1h进行预氧化处理;预氧化结束后,将气体氛围改为氩气氛围,流速设置为100ml/min,继续利用管式炉以10℃/min的升温速率升温至1100℃,并恒温2h进行高温热驱动处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,即制得ni-vc saa电催化co2还原材料。
[0065]
按照实施例1中的方法测试ni-vc saa电催化co2还原材料的催化性能。
[0066]
图5为ni-vc saa电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所述催化剂在不同电压下的产物法拉第效率数值,如图5所示。在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率仅为68.2~86.4%,当电压为-0.98v时,产物一氧化碳的法拉第效率达到最优值,为86.4%。电压降低(-0.78v),所述催化剂不能够有效地抑制析氢竞争反应,产物一氧化碳法拉第效率仅为68.2%,竞争产物氢气法拉第效率达到31.8%。
[0067]
同样的,参照实施例1,仅改变金属盐用量,分别替换为0.1、0.8、1.2mmol氯化镍,相应制得的电催化材料的催化性能如表2所示。
[0068]
表2
[0069]
氯化镍用量(mmol)ni:v摩尔比法拉第效率(-0.78~-1.18v)0.2(实施例3)0.5:168.2~86.4%0.4(实施例1)1:198.2~99.6%0.82:140.4~48.4%1.23:132.8~40.5%
[0070]
对比例1:
[0071]
取0.4mmol氯化镍和4g聚乙烯吡咯烷酮加入到20ml n,n-二甲基甲酰胺溶液中,在室温条件下,通过磁力搅拌12h制得均一透明的纺丝溶液,然后利用静电纺丝技术对该溶液进行纺丝,设置纺丝正极电压为18kv,接收器于纺丝针头间距为20cm,溶液流速为0.2ml/min,制得含有镍盐的纳米纤维膜。
[0072]
将上述纳米纤维膜裁剪为5cm
×
2cm的矩形,随后将其夹于石墨片中,置于管式炉的中间部位。在空气氛围下,先以10℃/min的升温速率升温至220℃,随后恒温1h进行预氧化处理;预氧化结束后,将气体氛围改为氩气氛围,流速设置为100ml/min,继续利用管式炉以10℃/min的升温速率升温至1100℃,并恒温2h进行高温热驱动处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,制得ni纳米颗粒催化剂;再经浓度为30%的硝酸进行酸洗,即制得ni sa电催化co2还原材料。
[0073]
按照实施例1中的方法测试ni-vc saa电催化co2还原材料的催化性能。
[0074]
图6为ni sa电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所
述催化剂在不同电压下的产物法拉第效率数值,如图6所示。ni sa电催化co2还原材料在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率仅为40.4~51.2%,竞争产物氢气法拉第效率较高,表明所述ni sa电催化co2还原材料的活性及一氧化碳选择性较差。
[0075]
同样的,参照实施例1,省略钒盐,并将氯化镍的用量调整至0.8mmol,其他不变,得到相应的催化材料。其催化性能结果为:在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率为59.4~68.6%。
[0076]
对比例2:
[0077]
取0.4mmol乙酰丙酮钒和4g聚乙烯吡咯烷酮加入到20ml n,n-二甲基甲酰胺溶液中,在室温条件下,通过磁力搅拌12h制得均一透明的纺丝溶液,然后利用静电纺丝技术对该溶液进行纺丝,设置纺丝正极电压为18kv,接收器于纺丝针头间距为20cm,溶液流速为0.2ml/min,制得含有镍盐与钒盐的纳米纤维膜。
[0078]
将上述纳米纤维膜裁剪为5cm
×
2cm的矩形,随后将其夹于石墨片中,置于管式炉的中间部位。在空气氛围下,先以10℃/min的升温速率升温至220℃,随后恒温1h进行预氧化处理;预氧化结束后,将气体氛围改为氩气氛围,流速设置为100ml/min,继续利用管式炉以10℃/min的升温速率升温至1100℃,并恒温2h进行高温热驱动处理;恒温结束后在惰性气体氛围下将管式炉自然冷却至室温,即制得vc电催化co2还原材料。
[0079]
按照实施例1中的方法测试ni-vc saa电催化co2还原材料的催化性能。
[0080]
图7为vc电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所述催化剂在不同电压下的产物法拉第效率数值,如图7所示。在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率仅为5.6~9.4%,所述催化剂不能够有效地抑制析氢竞争反应,竞争产物氢气法拉第效率超过90%。总体来看,所述催化剂表现出较差的二氧化碳还原活性及一氧化碳选择性。
[0081]
同样的,参照实施例1,省略镍盐,并将乙酰丙酮钒的用量调整至0.8mmol,其他不变,得到相应的催化材料。其催化性能结果为:在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率为8.2~10.6%。
[0082]
对比例3:
[0083]
用天平称量1.5mg对比例1中所制备的ni sa和1.5mg对比例2中所制备的vc催化剂,利用研磨机将其磨碎并混合均匀,然后将其分散于2ml乙醇中。随后将100μl nafion 117溶液加入至上述溶液中,超声30min。待溶液分散均匀后,利用喷枪将上述溶液均匀喷涂于碳纸(2
×
2cm2)上,将其置于红外灯下进行溶剂挥发,即制得ni sa-vc工作电极。
[0084]
按照实施例1中的方法测试ni-vc saa电催化co2还原材料的催化性能。
[0085]
图8为ni sa-vc电催化co2还原材料的产物法拉第效率图。通过相关公式计算得到所述催化剂在不同电压下的产物法拉第效率数值,如图8所示。在电压范围(-0.78~-1.18v vs.rhe)下,所述催化剂的产物一氧化碳法拉第效率仅为16.6~26.4%,所述催化剂不能够有效地抑制析氢竞争反应,竞争产物氢气法拉第效率超过70%。总体来看,所述催化剂表现出比实施例1差的二氧化碳还原活性及一氧化碳选择性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1