一种石菖蒲提取物中成分的检测方法与流程
本发明涉及一种石菖蒲提取物中成分的检测方法。
背景技术:
石菖蒲为天南星科菖蒲属植物,是一种常用中药,其根茎入药。《名医别录》记载:“久服聪耳明目,益心智,高智不老”。近年来药理报道,石菖蒲对小鼠学习和记忆有促进作用,对东莨菪碱造成的记忆获得障碍有明显的改善,可望治疗老年痴呆症。
石菖蒲的活性部位含有乙酰胺、胍基钠、钾或镁盐等和胍基甲酸等含氮类化合物,结构式如下,但是石菖蒲中还含有的细辛醚(α-细辛醚和β-细辛醚)、黄樟醚等有毒成分,有致癌、致畸、致突变等副作用。
在现有的报道中,有很多检测细辛醚或石菖蒲活性部位的方法。但是,目前缺少一种能够同时检测石菖蒲提取物中含氮化合物和有毒成分的检测方法。
技术实现要素:
本发明提供了一种石菖蒲提取物中同时检测含氮类化合物和毒性成分的检测方法。通过本发明的检测方法,可以同时测定石菖蒲提取物中的含氮类化合物和细辛醚,从而控制石菖蒲提取物的质量。
本发明提供了一种石菖蒲提取物中成分的检测方法,其包含如下步骤:将石菖蒲提取物经液相色谱检测,即可;其中,液相色谱的参数如下:填充 剂为十八烷基硅烷键合硅胶;流动相为甲醇和磷酸二氢钾水溶液,其中所述的磷酸二氢钾水溶液的pH为3.5~4.5,浓度为4.5~5.5mmol/L;梯度洗脱。
其中石菖蒲提取物可为本领域常规的以石菖蒲药材为原料进行常规方法提取而获得的石菖蒲提取物,本发明优选采用以下石菖蒲提取物的制备方法获得:将经水提醇沉的石菖蒲药材提取物的上清液,用大孔吸附树脂吸附,洗脱树脂,得洗脱液,即可。
其中,所述的石菖蒲药材为本领域常规用来制备石菖蒲提取物的石菖蒲药材。所述水提醇沉的方法和条件可参照本领域常规提取石菖蒲提取物的方法和条件。较佳地,将石菖蒲药材和水混合提取后,收集提取液;向所述提取液中加醇类溶剂,沉降,收集上清液,即为本发明所述的经水提醇沉的石菖蒲药材提取物的上清液。本发明特别优选以下条件:在所述水提的过程中,水的用量较佳地为石菖蒲质量的6~10倍,更佳地为6~8倍,最佳地为8倍;所述的水提的提取温度较佳地为90℃~100℃;所述的水提的提取时间较佳地为30~120分钟,更佳地为60~120分钟;所述的水提的提取次数较佳地为1~4次,更佳地为2~3次,最佳地为3次。
较佳地,在所述的醇沉之前,将提取液按本领域常规的浓缩方法进行浓缩,如减压浓缩;较佳地,将所述的提取液浓缩至1mL/1g石菖蒲药材~2mL/1g石菖蒲药材为止,更佳地浓缩至1mL/1g石菖蒲药材,收集浓缩液。
在所述的醇沉的过程中,所述的醇类溶剂为本领域常规的石菖蒲水提醇沉所用醇类溶剂,较佳地为乙醇;所述的乙醇较佳地为95%乙醇,百分数为体积百分数;所述的醇类溶剂的加入量较佳地为在所述的醇类溶剂与所述的浓缩液的混合溶液中,加入至含有65~80%的醇类溶剂,更佳地为含有65~75%的醇类溶剂,百分数均为体积百分数;所述的沉降的时间较佳地为加入醇类溶剂后沉降8~12小时;所述的上清液的收集方法可参照本领域常规方法,如:离心收集上清液。
在本发明所述的石菖蒲提取物的制备方法中,较佳地,将所述的上清液浓缩至干并和水混合溶解后,再用大孔吸附树脂吸附。所述的浓缩可按本领 域常规的浓缩方法进行操作,如减压浓缩;所述的水的用量为0.2mL/1g石菖蒲药材~0.5mL/1g石菖蒲药材。
所述的用大孔吸附树脂吸附可参照本领域常规的树脂吸附方法,本发明特别优选以下条件:所述的大孔吸附树脂较佳地为D101大孔吸附树脂,更佳地为D101大孔吸附树脂柱;所述的D101大孔吸附树脂柱的径高比较佳地为1∶4~1∶12,更佳地为1∶8~1∶12;所述的用大孔吸附树脂吸附的上样流速较佳地为1~2BV/h,更佳地为1~1.5BV/h。所述的大孔吸附树脂的用量较佳地为0.5mL树脂/1g石菖蒲药材~1mL树脂/1g石菖蒲药材,更佳地为0.6mL树脂/1g石菖蒲药材。
所述洗脱树脂的方法和条件可参照本领域常规树脂洗脱的方法和条件,较佳地,用树脂洗脱剂洗脱吸附后的大孔吸附树脂,收集树脂洗脱液,即可。本发明特别优选以下条件:所述的树脂洗脱剂较佳地为水,所述树脂洗脱剂的用量较佳地为4~6倍树脂体积,更佳地为5倍树脂体积。
本发明所述的石菖蒲提取物制备方法较佳地还包括活性炭吸附步骤:将所述树脂洗脱液用活性炭吸附,洗脱活性炭,收集活性炭洗脱液,干燥,即可。
较佳地,在所述的树脂洗脱液用活性炭吸附之前,将所述的树脂洗脱液按本领域常规的浓缩方法进行浓缩,如减压浓缩。较佳地,将树脂洗脱液浓缩至0.2mL/1g石菖蒲药材~0.5mL/1g石菖蒲药材为止。
所述的用活性炭吸附的方法和条件可参照本领域常规活性炭吸附的方法,本发明优选以下条件:所述的活性炭较佳地为活性炭柱,径高比较佳地为1∶4~1∶12,更佳地为1∶8~1∶12;所述的活性炭吸附的上样流速较佳地为1~2BV/h,更佳地为1~1.5BV/h。活性炭的用量较佳地为0.5mL活性炭/1g石菖蒲药材~1mL活性炭/1g石菖蒲药材,更佳地为0.6mL活性炭/1g石菖蒲药材。
所述的洗脱活性炭的方法和条件可参照本领域常规洗脱活性炭的方法和条件,较佳地,用不同活性炭洗脱剂依次洗脱吸附后的活性炭,收集活性 炭洗脱液,即可。本发明特别优选以下条件:所述的不同活性炭洗脱剂较佳地为水和乙醇水溶液;所述的乙醇水溶液较佳地为体积分数为30%的乙醇水溶液;所述的洗脱活性炭较佳地用所述的水洗脱活性炭后再用所述的乙醇水溶液洗脱活性炭;所述的水的用量较佳地为3~5倍活性炭体积,更佳地为4倍活性炭体积;所述的乙醇水溶液的用量较佳地为3~5倍活性炭体积,更佳地为4倍活性炭体积;所述的收集活性炭洗脱液较佳地收集30%乙醇水溶液的活性炭洗脱液。
所述的干燥可按本领域常规干燥方法操作,较佳地为减压浓缩后干燥。
本发明中,所述的磷酸二氢钾水溶液较佳地采用无机酸调节pH,更佳地采用磷酸调节pH;所述的磷酸二氢钾水溶液的pH较佳地为4.0;所述的磷酸二氢钾水溶液的浓度较佳地为5mmol/L。
本发明中,所述的梯度洗脱较佳地为:初始梯度为所述的磷酸二氢钾水溶液占所述的流动相的体积百分数为97%-100%,洗脱时间较佳地为15min;第二梯度为所述的磷酸二氢钾水溶液占所述的流动相的体积分数由97%-100%变化至75%,洗脱时间较佳地为10min;第三梯度为所述的磷酸二氢钾水溶液占所述的流动相的体积分数由75%变化至40%,洗脱时间较佳地为25min;第四梯度为所述的磷酸二氢钾水溶液占所述的流动相的体积分数由40%变化至20%,洗脱时间较佳地为10min;最终梯度为所述的磷酸二氢钾水溶液占所述的流动相的体积百分数为20%,洗脱时间较佳地为10-20min,更佳地为20min,百分数为体积百分数。
本发明中,所述液相色谱的参数还可包括:柱温较佳地为25-35℃,更佳地为30℃;所述的流动相的流速较佳地为0.4-0.6ml/min,更佳地为0.5ml/min;检测波长较佳地为210nm。
本发明较佳的色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以甲醇-5mmol/l磷酸二氢钾溶液(磷酸调pH4.0)为流动相;柱温30℃;流速0.5ml/min;检测波长分别为210nm;按表1或表2进行梯度洗脱。
表1
表2
本发明最佳的色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以甲醇-5mmol/l磷酸二氢钾溶液(磷酸调pH4.0)为流动相;柱温30℃;流速0.5ml/min;检测波长分别为210nm;按表1进行梯度洗脱。
在最佳色谱条件下获得的色谱图谱中,保留时间为6.8min的化合物为SCP-H-1,保留时间为10.3min的化合物为SCP-H-5,保留时间为10.8min的化合物为SCP-H-6,其结构式如下:
保留时间为63.5min的化合物为α-细辛醚,保留时间为62.0min的化合物为β-细辛醚。
本发明的检测方法的步骤可参照本领域常规的色谱检测步骤进行;发明优选以下步骤:
(1)对照品溶液的制备:取α-细辛醚,β-细辛醚对照品和SCP-H-1、SCP-H-5、SCP-H-6单体化合物对照品适量,精密称定,分别用甲醇制成每1ml含0.005mg对照品的溶液,即得;
(2)供试品溶液的制备:取石菖蒲提取物适量,精密称定,置容量瓶中,加水制成每1ml含5mg石菖蒲提取物的溶液,即得;
(3)测定方法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:本发明公开了一种检测方法,使用该方法可以同时测定石菖蒲提取物中的有效成分含氮类化合物和有毒成分α-细辛醚及β-细辛醚,从而控制石菖蒲提取物的质量。
附图说明
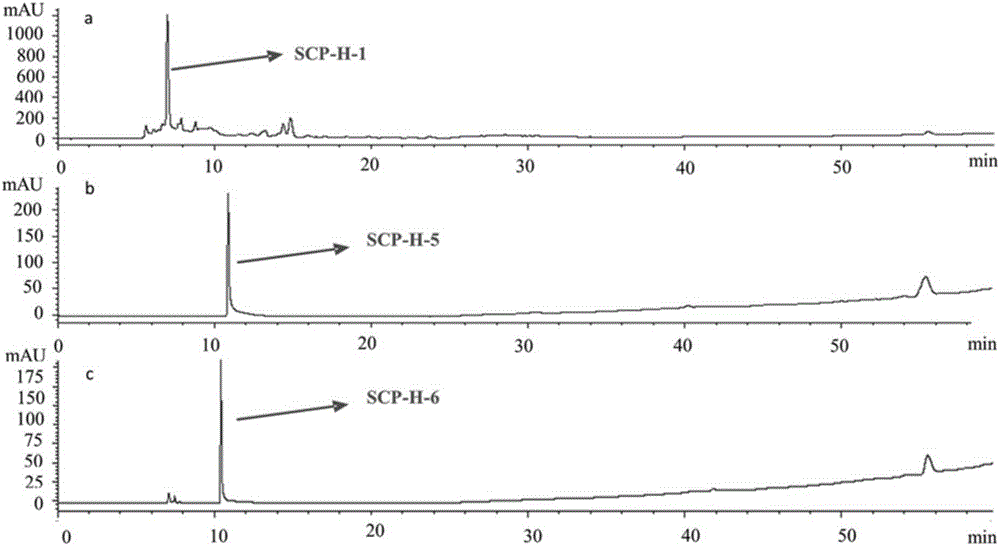
图1为采用实施例6的方法检测实施例6中所述的SCP-H-1、SCP-H-5和SCP-H-6对照品而获得的液相色谱图,其中,a为SCP-H-1的液相色谱图,b为SCP-H-5的液相色谱图,c为SCP-H-6的液相色谱图。
图2为采用实施例6的方法检测实施例6中所述的细辛醚对照品而获得的液相色谱图,谱图中保留时间62min左右的峰为β-细辛醚的保留峰,保留时间63.5min左右的峰为α-细辛醚的保留峰。
图3为采用实施例6的方法检测实施例6中所述的石菖蒲提取物而获得液相色谱图。
图4为采用实施例7的方法检测实施例7中所述的供试品溶液而获得的液相色谱图。
图5为采用对比例的方法检测对比例中所述的供试品溶液而获得的液相色谱图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1石菖蒲提取物的制备
取石菖蒲药材3.0kg,加入6倍量水,100℃下提取1小时,提取3次,过滤合并提取液,60℃减压浓缩至约3L,即浓缩至约1mL/g药材,加入95%乙醇至乙醇浓度为70%,沉降8h,离心取上清液,浓缩至无醇味,离心取上清液,浓缩至干,并加1000mL水使溶解,溶液上D101大孔吸附树脂,树脂用量为0.6mL树脂/1g石菖蒲药材,即1800mL树脂,柱径高比为1∶8,上样流速为1.0BV/h,上样结束后,用5BV去离子水洗脱树脂柱,收集水洗脱液,60℃减压浓缩至约600mL,冷却后上活性炭,活性炭柱用量为0.6mL活性炭/1g石菖蒲药材,即1800mL活性炭柱,柱径高比为1∶8,上样流速为1.0BV/h。上样结束后,先用4BV的去离子水洗脱活性炭柱,弃去水洗脱液,再用4BV的30%乙醇洗脱活性炭柱,收集30%乙醇洗脱液,60℃减压浓缩,干燥,即得样品20.62g,得率约0.69%。
实施例2石菖蒲提取物的制备
取石菖蒲药材5.0kg,加入8倍量水,100℃下提取1小时,提取3次,过滤合并提取液,60℃减压浓缩至约5L,即浓缩至1mL/1g药材,加入95%乙醇至乙醇浓度为70%,沉降12h,离心取上清液,浓缩至无醇味,离心取上清液,浓缩至干,并加1500mL水使溶解,溶液上D101大孔吸附树脂,树脂用量为0.6mL树脂/1g石菖蒲药材,即3000mL树脂柱,柱径高比为1∶8, 上样流速为1.0BV/h,上样结束后,用5BV去离子水洗脱树脂柱,收集水洗脱液,60℃减压浓缩至约1000mL,冷却后上活性炭,活性炭柱用量为0.6mL活性炭/1g石菖蒲药材,即3000mL,柱径高比为1∶8,上样流速为1.0BV/h。上样结束后,先用4BV的去离子水洗脱活性炭柱,弃去水洗脱液,再用4BV的30%乙醇洗脱活性炭柱,收集30%乙醇洗脱液,60℃减压浓缩,干燥,即得样品68.03g,得率约1.36%。
实施例3石菖蒲提取物的制备
取石菖蒲药材5.0kg,加入10倍量水,95℃下提取0.5小时,提取4次,过滤合并提取液,60℃减压浓缩至约10L,即浓缩至2mL/1g药材,加入95%乙醇至乙醇浓度为65%,沉降12h,离心取上清液,浓缩至无醇味,离心取上清液,浓缩至干,并加2500mL水使溶解,溶液上D101大孔吸附树脂,树脂用量为1mL树脂/1g石菖蒲药材,即5000mL树脂柱,柱径高比为1∶4,上样流速为2.0BV/h,上样结束后,用4BV去离子水洗脱树脂柱,收集水洗脱液,60℃减压浓缩至约2500mL,冷却后上活性炭,活性炭柱用量为1mL活性炭/1g石菖蒲药材,即5000mL,柱径高比为1∶4,上样流速为2.0BV/h。上样结束后,先用3BV的去离子水洗脱活性炭柱,弃去水洗脱液,再用3BV的30%乙醇洗脱活性炭柱,收集30%乙醇洗脱液,60℃减压浓缩,干燥,即得样品55.26g,得率约1.11%。
实施例4石菖蒲提取物的制备
取石菖蒲药材3.0kg,加入10倍量水,90℃下提取2小时,提取1次,过滤合并提取液,60℃减压浓缩至约6L,即浓缩至2mL/1g药材,加入95%乙醇至乙醇浓度为80%,沉降10h,离心取上清液,浓缩至无醇味,离心取上清液,浓缩至干,并加900mL水使溶解,溶液上D101大孔吸附树脂,树脂用量为0.5mL树脂/1g石菖蒲药材,即1500mL树脂柱,柱径高比为1∶12,上样流速为1.5BV/h,上样结束后,用6BV去离子水洗脱树脂柱,收集水洗脱液,60℃减压浓缩至约900mL,冷却后上活性炭,活性炭柱用量为0.5mL活性炭/1g石菖蒲药材,即1500mL,柱径高比为1∶12,上样流速为1.5BV/h。 上样结束后,先用5BV的去离子水洗脱活性炭柱,弃去水洗脱液,再用5BV的30%乙醇洗脱活性炭柱,收集30%乙醇洗脱液,60℃减压浓缩,干燥,即得样品29.18g,得率约0.97%。
实施例5石菖蒲提取物的制备
取石菖蒲药材3.0kg,加入6倍量水,100℃下提取2小时,提取2次,过滤合并提取液,60℃减压浓缩至约3L,即浓缩至1mL/1g药材,加入95%乙醇至乙醇浓度为75%,沉降12h,离心取上清液,浓缩至无醇味,离心取上清液,浓缩至干,并加600mL水使溶解,溶液上D101大孔吸附树脂,树脂用量为0.7mL树脂/1g石菖蒲药材,即2100mL树脂柱,柱径高比为1∶8,上样流速为1BV/h,上样结束后,用5BV去离子水洗脱树脂柱,收集水洗脱液,60℃减压浓缩至约1000mL,冷却后上活性炭,活性炭柱用量为0.7mL活性炭/1g石菖蒲药材,即2100mL,柱径高比为1∶8,上样流速为1BV/h。上样结束后,先用5BV的去离子水洗脱活性炭柱,弃去水洗脱液,再用5BV的30%乙醇洗脱活性炭柱,收集30%乙醇洗脱液,60℃减压浓缩,干燥,即得样品30.09g,得率约1.00%。
实施例6石菖蒲提取物的检测
检测条件
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以甲醇-5mmol/l磷酸二氢钾溶液(磷酸调pH4.0)为流动相;柱温30℃;流速0.5ml/min;检测波长分别为210nm;按表3进行梯度洗脱。
表3
对照品溶液的制备:取α-细辛醚,β-细辛醚对照品和SCP-H-1、SCP-H-5、SCP-H-6单体化合物对照品适量,精密称定,分别用甲醇制成每1ml含0.005mg对照品的溶液,即得。
供试品溶液的制备:取实施例2制备的石菖蒲提取物适量,精密称定,置量瓶中,加水制成每1ml含5mg石菖蒲提取物的溶液,即得。
测定方法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
获得对照品的液相色谱图如图1和图2所示;获得供试品的液相色谱图谱如图3所示。
在该色谱条件下,由图1可知,化合物SCP-H-1的保留时间为6.8min(a),化合物SCP-H-5的保留时间为10.3min(b),化合物SCP-H-6的保留时间为10.8min(c);由图2可知,α-细辛醚的保留时间为63.5min,β-细辛醚的保留时间为62.0min。
图3中,在保留时间6min、7min和8min左右出现了保留峰,表明实施例2制备的石菖蒲提取物主要含有SCP-H-1,并且在SCP-H-1的影响下,SCP-H-5和SCP-H-6的保留峰出现前移现象。在实施例2的提取物中未发现毒成分细辛醚。在图1、图2和图3中,溶剂峰在洗脱梯度变化时均有不同程度的出现,但均不明显。
实施例7:
检测条件
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以甲醇-5mmol/l磷酸二氢钾溶液(磷酸调pH4.0)为流动相;柱温30℃;流速0.5ml/min;检测波长为210nm;按表4进行梯度洗脱。
表4
供试品溶液的制备:取α-细辛醚,β-细辛醚和SCP-H-1、SCP-H-5单体化合物适量,精密称定后按照四种供试品的质量比为1∶1∶1∶1混合,用甲醇制成每1ml含0.005mg供试品的溶液,即得。
测定方法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
获得液相色谱图谱如图4所示。图4中,在保留时间6min和7min左右以及62min和64min左右出现了保留峰,对应石菖蒲活性部位SCP-H-1和SCP-H-5以及β-细辛醚和α-细辛醚,表明本发明的检测方法能够同时检测出石菖蒲活性部位和细辛醚。在图4中,溶剂峰在洗脱梯度变化时均有不同程度的出现,但均不明显。
对比例:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;流动相为甲醇-水(6∶4),1000ml中加入磷酸二氢钾1.4g,庚烷磺酸钠1.2g;柱温30℃;流速1.0ml/min;检测波长为257nm。
供试品溶液的制备:取α-细辛醚,β-细辛醚对照品和SCP-H-1、SCP-H-5、单体化合物适量,精密称定,用甲醇制成每1ml含0.005mg的溶液,即得。
获得液相色谱图谱如图5所示。图5中,石菖蒲活性部位和细辛醚的保留峰出现位置前移,α-细辛醚在12min左右出现保留峰,β-细辛醚在9min左右出现保留峰,而石菖蒲活性部位的保留峰与溶剂峰混在一起,无法分辨出来,在这种情况下,无法检测到SCP-H-1、SCP-H-5。因此,对比例中的检测方法不能同时检测出石菖蒲活性部位和细辛醚。
- 还没有人留言评论。精彩留言会获得点赞!