用于细胞成像和药物筛选的基于具有聚集诱导发射特征之荧光团的发光探针的制作方法
本申请要求于2014年1月27日提交的美国临时申请No.61/932,007的权益。上述申请的全部教导均通过引用并入本文。
背景技术:
细胞内分子监测和药物筛选可提供对细胞的生物学状态和药物的治疗效力的有价值了解。已使用基于聚集诱导发射(aggregation-induced emission,AIE)荧光团和水溶性肽的荧光发光探针(light-up probe)来进行细胞蛋白质的实时监测。第一代特异性AIE探针的设计受限于具有高水溶性的基于肽的识别元件。为了扩展设计原理以包括更广泛的识别元件(例如,如疏水性分子、小分子等),有必要开发更通用的策略。
发明概述
本发明涉及AIE荧光团(包括具有激发态分子内质子转移(excited state intramolecular proton transfer,ESIPT)特征的那些)、AIE发光探针的开发,以及它他们在感测、成像和药物筛选中的应用。
更特别地,本文中描述了一系列的荧光发光探针,其一般性地包含AIE荧光团、识别部分、靶向配体和亲水单元(例如,5个天冬氨酸)以确保该探针的良好水溶性。由于AIE荧光团的独特性质,所述探针在水性介质中是非荧光的,但是当被细胞内分子切割后变得具有高发射性。所述探针能够以高信噪比实现细胞内分子的发光监测和药物筛选。基于类似的设计原理,用表现出AIE和ESIPT特征二者的荧光团取代传统的AIE荧光团可简化这样的设计,因为所述探针总是非荧光的而与探针的水溶性无关。由于AIE探针设计策略可通过简单地将识别部分替换成化学生物学中其他的可切割接头而推广用于执行多种任务,其开创了设计用于细胞内分子成像和药物筛选的特异性发光探针的新机会。
附图简述
如附图中所举例说明的,前述内容将根据对本发明的示例性实施方案的以下更具体描述而显而易见,在附图中类似的附图标记指在不同视图中的相同部分。附图不必按比例绘制,相反,重点在于对本发明的一些实施方案进行举例说明。
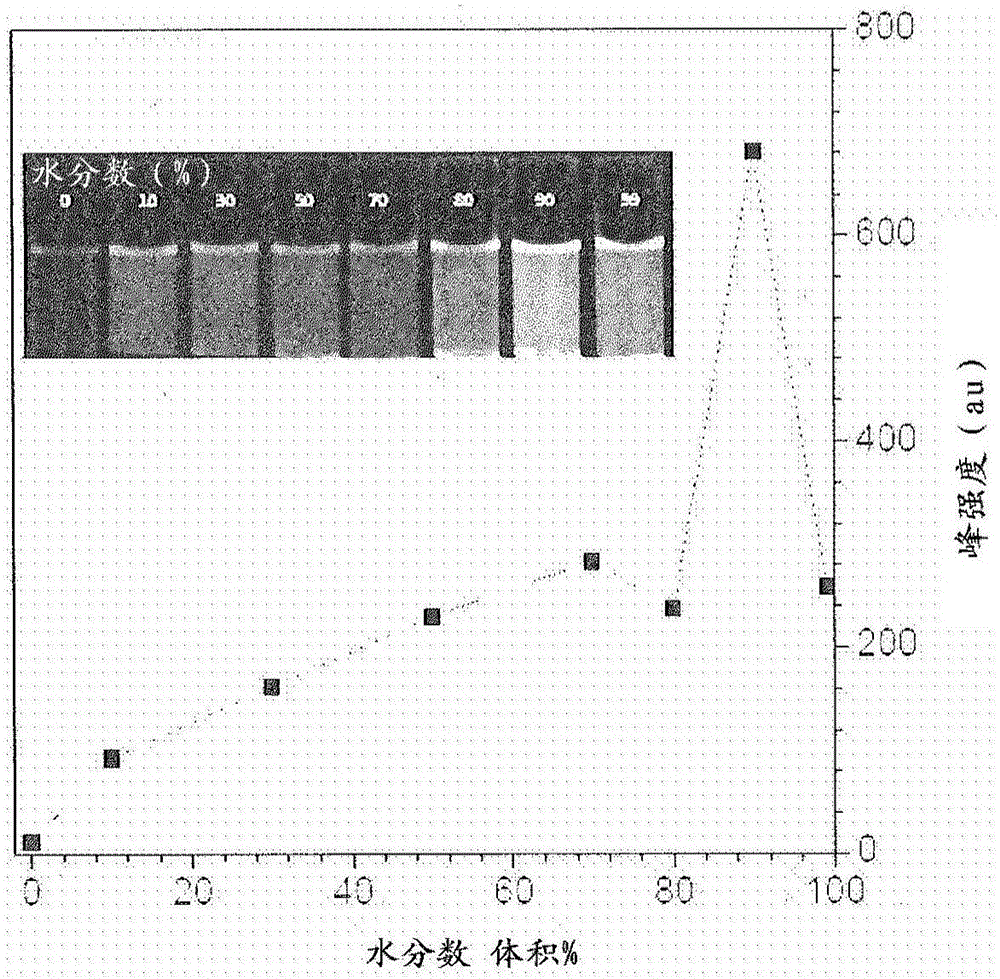
图1A至1B示出了DMA-HC的发射谱和峰强度图。图1A示出了在具有不同水分数(fw)的THF/水混合物中DMA-HC(20μM)的发射谱。图1B示出了峰强度相对于fH·λex=430nm的图。
图2A至2B示出了HC-phos的发射谱和比率荧光(ratiometric fluorescence)强度图。图2A示出了40μM HC-phos在37℃下在添加不同活性的ALP之后在50mM Tris-HCl缓冲溶液(pH 9.2)中的发射谱。图2B示出了比率荧光强度(I641/I539)相对于ALP活性(孵育时间:45分钟;λex=430nm)的图。
图3示出了经孵育25分钟的Hela细胞的明场和荧光图像(激发波长为460nm至490nm)。图像A至B分别示出了未经染色之对照的明场和荧光图像。图像C至D分别示出了经HC-phos(10μM)处理的细胞的明场和荧光图像。图像E至F分别示出了经HC-phos(10μM)和左旋咪唑(4mM)处理的细胞的明场和荧光图像。
图4示出了显示二乙基二硫代氨基甲酸盐(DDTC)与顺铂和还原的Pt(IV)前药之反应的HPLC色谱图。色谱图A示出了单独的DDTC。色谱B示出了DDTC与顺铂的反应。色谱图C示出了DDTC与TPS-DEVD-Pt-cRGD的反应。色谱图D示出了DDTC与TPS-DEVD-Pt-cRGD(10μM)在1mM抗坏血酸存在下持续12小时的反应。
图5A至5F示出了TPS-CH2N3、TPS-DEVD-NH2和TPS-DEVD-Pt-cRGD的光致发光(photoluminescence,PL)谱和/或图。图5A示出了TPS-CH2N3、TPS-DEVD-NH2、TPS-DEVD-Pt-cRGD在DMSO/PIPE(v/v=1/199)中的PL谱。图5A中的摄影插图拍摄于UV灯在365nm处的照明之下。图5B示出了TPS-DEVD-Pt-cRGD在经抗坏血酸和胱天蛋白酶-3于抑制剂5-[(S)-(+)-2-(甲氧基甲基)吡咯烷基]磺酰基靛红(10μM)存在和不存在下处理之后的PL谱。图5C示出了TPS-DEVD-Pt-cRGD在经抗坏血酸处理之后在DMSO/PIPE缓冲液(v/v=1/199)中在胱天蛋白酶-3存在下的时间依赖性荧光谱。图5D示出了经抗坏血酸(1mM)预处理的TPS-DEVD-Pt-cRGD(10μM)在添加在胱天蛋白酶-3(200pM)之后从0至120分钟在485nm处的PL强度。图5D的数据表示平均值+/-标准偏差,n=3。图5E示出了经抗坏血酸(1mM)预处理的TPS-DEVD-Pt-cRGD(10μM)与不同量的胱天蛋白酶-3(0、10、25、50、100和200pM)在DMSO/PIPE缓冲液(v/v=1/199)中孵育60分钟的PL谱。图5F示出了经抗坏血酸(1mM)预处理的TPS-DEVD-Pt-cRGD(10μM)在DMSO/PIPE缓冲液(v/v=1/199)中在添加不同量胱天蛋白酶-3保持60分钟之后在485nm处的PL强度。图5F的数据表示平均值+/-标准偏差,n=3。
图6A至6B示出了TPS-DEVD-Pt-cRGD的(I-I0)/I0图和PL谱。图6A示出了TPS-DEVD-Pt-cRGD在与不同的蛋白质孵育60分钟之后的(I-I0)/I0的图,其中I和I0分别是蛋白质浓度为200pM和0pM时的PL强度。图6B示出了TPS-DEVD-Pt-cRGD在凋亡U87-MG细胞裂解液(Ap-U87-MG)和正常U87-MG细胞裂解液(Nor-U87-MG)中的时间依赖性PL谱。图6B的数据表示平均值+/-标准偏差,n=3。
图7示出了涉及经TPS-DEVD-Pt-cRGD处理的细胞的CLSM和共聚焦图像以及荧光/核覆盖图像(所有的图像均共有20μm的相同比例尺)。图像A至D示出了展示经TPS-DEVD-Pt-cRGD(5μM)染色的U87-MG细胞的凋亡进程的实时CLSM图像(细胞核用DRAQ5进行活染色)。图像E至F分别示出了MCF-7和293T细胞在经TPS-DEVD-Pt-cRGD(5μM)处理6小时之后的共聚焦图像(细胞核用DRAQ5进行活染色)。图像G至L分别是图像A至F的对应荧光/核覆盖图像。
图8示出了U87-MG细胞在经TPS-DEVD-Pt-cRGD处理之后的CLSM图像(所有的图像均共有20μm的相同比例尺)。A部分的三幅图像示出了U87-MG细胞在经TPS-DEVD-Pt-cRGD(5μM)和胱天蛋白酶-3抗体于抑制剂不存在下处理之后的CLSM图像。B部分的三幅图像示出了U87-MG细胞在经TPS-DEVD-Pt-cRGD(5μM)和胱天蛋白酶-3抗体于抑制剂(5μM)存在下处理之后的CLSM图像。
图9A至9B示出了细胞生存力和凋亡引起的PL强度的相关性。图9A示出了U87-MG细胞在经不同浓度的TPS-DEVD-Pt-cRGD处理之后的细胞生存力(72小时)和凋亡引起的PL强度(6小时)的相关性。图9B示出了MCF-7细胞在经不同浓度的TPS-DEVD-Pt-cRGD处理之后的细胞生存力(72小时)和凋亡引起的PL强度(6小时)的相关性。
图10A至10D示出了HPLC色谱图以及PL谱和PL强度图。图10A示出了举例说明DDTC与顺铂和还原的Pt(IV)前药之反应的HPLC色谱图:(1)单独的DDTC、(2)DDTC与顺铂的反应、(3)DDTC与PyTPE-Pt-D5-cRGD的反应、(4)DDTC与PyTPE-Pt-D5-cRGD(10μM)在抗坏血酸(1mM)存在下的反应,该反应持续12小时。图10B示出了PyTPE-NH2和PyTPE-Pt-D5-cRGD在DMSO/PBS混合物(v/v=1/199)中的PL谱。图10B中的摄影插图拍摄于UV灯在365nm处的照明之下。图10C示出了经抗坏血酸(1mM)处理的PyTPE-Pt-D5-cRGD(10μM)的时间依赖性PL谱。图10D示出了与抗坏血酸(1mM)在DMSO/PBS(v/v=1/199)中孵育的PyTPE-Pt-D5-cRGD在605nm处的PL强度相对于浓度的曲线。图10D的数据表示平均值+/-标准偏差,n=3。
图11示出了MDA-MB-231和MCF-7细胞在孵育之后的共聚焦显微术图像(所有的图像均共有20μm的相同比例尺)。图像A示出了MDA-MB-231细胞在与PyTPE-Pt-D5-cRGD孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。图B示出了MDA-MB-231细胞在与PyTPE-Pt-D5孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。图C示出了MDA-MB-231细胞在与PyTPE-C6-D5-cRGD孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。图像D示出了MCF-7细胞在与PyTPE-Pt-D5-cRGD孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。图像E示出了MCF-7细胞在与PyTPE-Pt-D5孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。图像F示出了MCF-7细胞在与PyTPE-C6-D5-cRGD孵育之后的共聚焦图像(细胞核用Hoechst 33342进行染色)。
图12A至12B示出了MDA-MB-231和MCF-7细胞在处理之后的细胞生存力。图12A示出了MDA-MB-231细胞在经不同浓度的PyTPE-Pt-D5-cRGD、PyTPE-Pt-D5和PyTPE-C6-D5-cRGD处理72小时之后的细胞生存力。图12B示出了MCF-7细胞在经不同浓度的PyTPE-Pt-D5-cRGD、PyTPE-Pt-D5和PyTPE-C6-D5-cRGD处理72小时之后的细胞生存力。
图13A至13D示出了PL谱以及PL强度图。图13A示出了TPE-CH2NH2和TPE-SS-D5-cRGD在DMSO/PBS(v/v=1/199)中的PL谱。图13A中的摄影插图拍摄于UV灯的照明之下。图13B示出了经GSH处理的TPE-SS-D5-cRGD的时间依赖性PL谱。图13C示出了TPE-SS-D5-cRGD(1.0mM)在不同浓度的GSH存在下的PL谱。图13D示出了在470nm下的PL强度相对于GSH浓度的图(平均值+/-标准偏差,n=3)。
图14示出了U87-MG和MCF-7细胞在孵育之后的共聚焦显微术图像(所有的图像均共有20μm的相同比例尺)。图像A示出了U87-MG细胞在与TPE-SS-D5-cRGD孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。图像B示出了U87-MG细胞在与TPE-SS-D5孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。图像C示出了U87-MG细胞在与TPE-CC-D5孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。图像D示出了MCF-7细胞在与TPE-SS-D5-cRGD孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。图像E示出了MCF-7细胞在与TPE-SS-D5孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。图像F示出了MCF-7细胞在与TPE-CC-D5孵育之后的共聚焦图像(细胞核用碘化丙啶进行染色)。
发明详述
本发明涉及发光团(luminogen)(为其亚类的AIE荧光团和AIE-ESIPT荧光团)以及包含靶标识别基序、亲水性部分、连接部分和至少一个发光团的化学组合物(例如,发光探针)。本发明还涉及使用包含发光团的所述组合物来评估前药转变成其活性形式、评估前药的治疗效力、检测生物样品中的谷胱甘肽、检测样品中的碱性磷酸酶以及进行荧光成像或核磁共振成像的方法。
定义
下文所述的取代基的所有定义还适用于这些术语与另外取代基的联合使用。
“烷基”意指饱和的脂肪族支链或直链单价烃基,通常C1-C10,优选C1-C6。“(C1-C6)烷基”意指线性或分支排列的具有1至6个碳原子的基团。“(C1-C6)烷基”包括甲基、乙基、丙基、丁基、叔丁基、戊基和己基。
“亚烷基”意指饱和的脂肪族直链二价烃基。因此,“(C1-C6)亚烷基”意指线性排列的具有1至6个碳原子的二价饱和脂肪族基团。“(C1-C6)亚烷基”包括亚甲基、亚乙基、亚丙基、亚丁基、亚戊基和亚己基。
“环烷基”意指饱和的脂肪族环状烃环。因此,“C3-C8环烷基”意指(3至8元)饱和的脂肪族环状烃环。C3-C8环烷基包括但不限于环丙基、环丁基、环戊基、环己基、环庚基和环辛基。优选地,环烷基是C3-C6环烷基。
术语“烷氧基”意指-O-烷基;“羟基烷基”意指经羟基取代的烷基;“芳烷基”意指经芳基取代的烷基;“烷氧基烷基”意指经烷氧基取代的烷基;“烷基胺”意指经烷基取代的胺;“环烷基烷基”意指经环烷基取代的烷基;“二烷基胺”意指经两个烷基取代的胺;“烷基羰基”意指-C(O)-A*,其中A*是烷基;“烷氧基羰基”意指C(O)OA*,其中A*是烷基;并且此处烷基如上定义。烷氧基优选地是O(C1-C6)烷基,并且包括甲氧基、乙氧基、丙氧基、丁氧基、戊氧基和己氧基。
“环烷氧基”意指-O-环烷基,其中环烷基如上定义。示例性的(C3-C7)环烷氧基包括环丙氧基、环丁氧基、环戊氧基、环己氧基和环庚氧基。
单独使用或者作为较大部分如“芳基烷基”、“芳基烷氧基”、“芳基氧基”或“芳基氧基烷基”中的一部分使用的术语“芳基”意指碳环芳族环。术语“碳环芳族基团”可与术语“芳基”、“芳基环”、“碳环芳族环”、“芳基基团”和“碳环芳香基团”互换使用。芳基通常具有6至16个环原子。“经取代的芳基”在任意一个或更多个可取代的环原子处被取代。本文中使用的术语“C6-C16芳基”意指含有6至16个碳原子的单环的、双环的或三环的碳环环体系,并且包括苯基(Ph)、萘基、蒽基、1,2-二氢萘基、1,2,3,4-四氢萘基、芴基、茚满基(indanyl)、茚基等。在一些具体实施方案中,芳基是(C6-C10)芳基。(C6-C10)芳基(C1-C6)烷基通过(C6-C10)芳基(C1-C6)烷基中的(C1-C6)烷基部分与分子的剩余部分连接。
“杂”指环体系中的至少一个碳原子成员被选自N、S和O的至少一个杂原子替换。所述杂原子可任选地携带电荷。当N是环体系的杂原子时,其可另外地被包括以下的一个或更多个取代基取代:H、OH、O-、烷基、芳基、杂环基、环烷基或亚烯基(alkenylene),其中任意的烷基、芳基、杂环基、环烷基或亚烯基可任选地并且独立地被选自卤素、氰基、硝基、羟基、磷酸根(PO43-)或磺酸根(SO3-)的一个或更多个取代基取代。
“杂环”意指饱和的或部分不饱和的(3至7元)含有一个氮原子并且任选地另外含有1个独立地选自N、O或S的杂原子的单环杂环。当一个杂原子是S时,其可任选地是单或二氧化的(-S(O)-或S(O)2)。单环杂环的实例包括但不限于氮杂环丁烷、吡咯烷、哌啶、哌嗪、六氢嘧啶、四氢呋喃、四氢吡喃、吗啉、硫代吗啉、硫代吗啉1,1-二氧化物、四氢-2H-1,2-噻嗪、四氢-2H-1,2-噻嗪1,1-二氧化物、异噻唑烷或异噻唑烷1,1-二氧化物。杂环可任选地与如例如吲哚中的碳环稠合。
单独使用或者作为较大部分如“杂芳基烷基”或“杂芳基烷氧基”中的一部分使用的术语“杂芳基”、“杂芳族”、“杂芳基环”、“杂芳基基团”和“杂芳族基团”指具有总共5至14个环原子的芳族环基团,所述环原子选自碳和至少1个(通常1至4个,更通常1或2个)杂原子(例如,氧、氮或硫)。其包括单环和其中单环杂芳族环与一个或更多个其他碳环芳族环或杂芳族环稠合的多环。本文中使用的术语“5至14元杂芳基”意指含有一个或两个芳族环的单环的、双环的或三环的环体系,并且除非另外指出,否则在其总共5至14个原子中,1、2、3、4或5个是独立地选自N、NH、N(C1-6烷基)、O和S的杂原子。(C3-C10)杂芳基包括呋喃基、苯硫基、吡啶基、吡咯基、咪唑基,并且在本发明的一些优选实施方案中,杂芳基是(C3-C10)杂芳基。
“卤素”和“卤代”在本文中可互换使用,并且各自指氟、氯、溴或碘。
“氰基”意指-C≡N。
“硝基”意指-NO2。
本文中使用的氨基可以是伯氨基(NH2)、仲氨基(NHRx)或叔氨基(NRxRy),其中Rx和Ry可以是任意烷基、芳基、杂环基、环烷基或亚烯基,其各自任选地并且独立地被上述一个或更多个取代基取代。Rx和Ry取代基可一起形成“环”,其中该“环”(如本文中所用)是环状氨基(例如哌啶和吡咯烷)并且可含有杂原子,例如吗啉中。
术语“卤代烷基”、“卤代环烷基”和“卤代烷氧基”意指根据情况可被一个或更多个卤素原子取代的烷基、环烷基或烷氧基。术语“卤素”意指F、Cl、Br或I。
术语“酰基”意指-C(O)A*,其中A*是任选经取代的烷基或芳基(例如,任选经取代的苯基)。
“亚烷基”由-[CH2]z表示,其中z是正整数,优选1至8,更优选1至4。
“亚烯基”是其中至少一对相邻亚甲基被-CH=CH替换的亚烷基。
术语苄基(Bn)指-CH2Ph。
术语“烯基”意指包含至少一个双键的直链或支链烃基。(C6-C10)芳基(C2-C6)烯基通过(C6-C10)芳基(C2-C6)烯基中的(C2-C6)烯基部分与分子的剩余部分连接。
还包括本发明化合物的可药用盐。例如,含有胺或其他碱性基团的本发明化合物的酸式盐可通过使该化合物与合适的有机酸或无机酸反应产生可药用的阴离子盐形式来获得。阴离子盐的实例包括醋酸盐、苯磺酸盐、苯甲酸盐、碳酸氢盐、酒石酸氢盐、溴化物、依地酸钙、樟脑磺酸盐、碳酸盐、氯化物、柠檬酸盐、二盐酸盐、依地酸盐、乙二磺酸盐(edisylate)、丙酸酯十二烷基硫酸盐(estolate)、乙磺酸盐、富马酸盐、葡庚糖酸盐(glyceptate)、葡萄糖酸盐、谷氨酸盐、乙醇酰阿散酸盐、己基间苯二酚盐、氢溴酸盐、盐酸盐、羟基萘甲酸盐、碘化物、羟乙基磺酸盐、乳酸盐、乳糖酸盐、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐、甲基硫酸盐、粘酸盐(mucate)、萘磺酸盐、硝酸盐、扑酸盐、泛酸盐、磷酸盐/二磷酸盐、聚半乳糖醛酸盐、水杨酸盐、硬脂酸盐、碱式乙酸盐、琥珀酸盐、硫酸盐、鞣酸盐、酒石酸盐、氯茶碱盐(teoclate)、甲苯磺酸盐和三乙基碘盐
含有羧酸或其他酸性官能团的本发明化合物的盐可通过与合适的碱反应来制备。这样的可药用盐可用提供可药用阳离子的碱来制备,其包括碱金属盐(尤其是钠盐和钾盐)、碱土金属盐(尤其是钙盐和镁盐)、铝盐和铵盐,以及由诸如以下生理学上可接受的有机碱制成的盐:三甲胺、三乙胺、吗啉、吡啶、哌啶、皮考啉、二环己基胺、N,N’-二苄乙烯二胺、2-羟乙胺、双-(2-羟乙基)胺、三-(2-羟乙基)胺、普鲁卡因、二苄基哌啶、脱氢枞胺、N,N’-双脱氢枞胺、葡糖胺、N-甲基葡糖胺、可力丁、奎宁、喹啉和碱性氨基酸,例如赖氨酸和精氨酸。
本文中使用的术语“发光团”指展现出光发射的分子。如果所发射的光是荧光,则可替选地将该发光团称为荧光团。
“聚集诱导发射”指其中发光团当分散在例如有机溶剂中时发射光很少或者不发射光的特性。然而,在发光团分子例如以固体状态或者在水中聚集之后,由于发光团的疏水性,从发光团的光发射显著增强。
本文中使用的“靶标识别基序”是对生物靶标(例如蛋白质、肽或细胞膜中的受体)具有亲和力的化学部分。靶标识别基序可包含对特定靶结构具有亲和力的肽、蛋白质、寡核苷酸或有机官能团。
本文中使用的“连接部分”是将两个或更多个基团通过一个共价键或者通过一系列共价键连接的化学部分。连接部分的实例包括二硫键(disulfide)基团、氨基、2-硝基苄基衍生物、砜、腙、邻位二醇或者简单地一个或更多个共价键。连接部分的另一些实例可见于Bioorg.Med.Chem.,2012,20,571-582的表1中。
用于本发明组合物中的亲水性部分可包括水溶性聚合物或经带电侧基(side group)官能化的烷基链。用于本发明的水溶性聚合物的实例包括聚乙二醇或聚乙烯亚胺。可用于本发明的带电侧基包括例如SO33-或PO43-。
本文中使用的“光谱术”涵盖物质与辐射能反应的任何方法。这包括但不以任何方式限于显微术、荧光显微术、UV/Vis光谱测定法和流式细胞术。本文中使用的“微孔板读取仪”意指测量微孔板中包含的样品的例如荧光、吸光度和发光的实验室仪器。
本文中使用的“前药”是通常以其无活性形式施用于对象并且在该对象体内转变成其活性形式的治疗性化合物。例如,前药可包括铂(IV)[Pt(IV)]络合物,其转变为活性的铂(Ⅱ)[Pt(II)]络合物。在某些实施方案中,这样的转变通过用化学试剂还原发生。在某些其他实施方案中,这样的转变通过代谢过程发生。
四苯基乙烯或TPE是:
四苯基噻咯(tetraphenylsilole)或TPS是:
本文中使用的“活靶细胞”是作为治疗或治疗性方案之靶标的活细胞。在一些实施方案中,活靶细胞可以是作为前药之治疗靶标的癌细胞。
下面是对本发明的多个方面的描述。
发光团
在本发明的该第一方面,对发光团进行描述。发光团是发光的原子或发光原子的群。与发光团类似,荧光团是发荧光的原子或发荧光原子的群。发光是发射光的过程。发光的类型包括生物发光、化学发光、电致发光、电化学发光、光致发光等。已知一种类型的光致发光为荧光。因此,荧光团是发光团的一个子集。
AIE荧光团
在荧光团家族内,可发现聚集诱导发射(AIE)荧光团。本发明该方面的一个实施方案涉及具有下式结构的AIE荧光团或其可药用盐:
其中:
R1选自H、(C1-C6)烷基、(C3-C6)环烷基、(C6-C10)芳基、(C3-C10)杂芳基或(C2-C6)烯基;
R2独立地选自H、NHR3、N(R3)2、(C1-C6)烷基、(C3-C6)环烷基、(C6-C10)芳基、(C3-C10)杂芳基、-O(C1-C6)烷基、(C2-C6)烯基、CH=CH((C3-C10)杂芳基)或CH=CH((C6-C10)芳基);并且
R3选自H、(C1-C6)烷基或(C3-C6)环烷基。
AIE荧光团还任选地并且独立地被选自以下的一个或更多个取代基取代:
(C3-C10)杂芳基、其中*表示与发光团残基连接的点,并且**表示与前药、靶标识别基序或亲水性肽连接的点。
在另一个实施方案中,AIE荧光团具有下式的结构:
其中R1是(C1-C6)烷基。在一个优选实施方案中,R1是C2H5或C6H13。
在又一个实施方案中,AIE荧光团具有下式的结构:
以下方案对数种示例性AIE荧光团的设计和合成进行了更具体的举例说明。
示例性AIE荧光团的更详细合成途径可见于本申请的实施例部分。
AIE-ESIPT荧光团
在AIE荧光团家族内,可发现具有激发态分子内质子转移(ESIPT)特征的那些(AIE-ESIPT荧光团)。
本发明该方面的一个实施方案涉及具有下式结构的荧光团或其可药用盐:
其中:
M选自S、O或NH;
Q选自P(=O)(OH)2或C(O)O(C1-C6)烷基,其中C(O)O(C1-C6)烷基任选地被选自以下的一个或更多个取代基官能化:SH、OH、NH2或者任选地经选自OH、SH或NH2的一个或更多个取代基取代的(C6-C10)芳基;
R4选自NHR6、N(R6)2、(C1-C6)烷基、(C3-C6)环烷基、(C6-C10)芳基、(C3-C10)杂芳基、-O(C1-C6)烷基、-O(C3-C6)环烷基或(C2-C6)烯基;
R5是(C0-C6)烷基、其任选地被连接部分官能化;并且
R6选自H、(C1-C6)烷基或(C3-C6)环烷基。
在一个优选实施方案中,连接部分(如果存在的话)与靶标识别基序共价连接。靶标识别基序优选地对细胞膜受体具有亲和力。更优选地,靶标识别基序具有对整联蛋白αvβ3具有亲和力的环(Arg-Gly-Asp)(cRGD)残基。
在另一个实施方案中,AIE-ESIPT荧光团具有下式的结构:
其被称为Phos-HC。
以下方案对示例性AIE-ESIPT荧光团的设计和合成进行了更具体的举例说明。
该示例性AIE荧光团在检测样品之碱性磷酸酶中的用途可见于本申请的实施例部分。
AIE荧光团和AIE-ESIPT荧光团二者均可用于产生用于进行荧光成像和磁共振成像以及用于评估前药转变成其活性形式、评估前药的治疗效力、检测生物样品中的谷胱甘肽和检测样品中的碱性磷酸酶的发光探针。
发光探针
对个性化医疗而言高度期望以最小的副作用并实时原位监测药物效力来向肿瘤细胞递送靶向药物。更特别地,如果可设计并开发能够同时递送药物并非侵入性地原位评价治疗响应的系统,则是高度期望的。对该问题的最有希望解决方案是向系统内并入凋亡传感物(sensor)。
本发明的发明人已开发了基于包含具有AIE特征之荧光团的探针(发光探针)来实时监测体外和体内细胞凋亡的策略。将首先对发光探针的组合物进行讨论,之后对发光探针的应用进行描述。
在本发明的发光探针方面,发光探针是包含至少一个发光团、亲水性部分、连接部分和靶标识别基序的化学组合物,其中所述发光团表现出聚集诱导发射特性,并且进一步其中所述靶标识别基序、亲水性部分、连接部分和至少一个发光团通过共价连接以线性阵列连接。
在一个实施方案中,连接部分是前药,例如,如铂(IV)络合物(“Pt”)。
在另一个实施方案中,连接部分是可切割的连接基团。优选地,可切割的连接部分是二硫键(“SS”)。或者,所述可切割的连接基团可以是可以在酸性条件中被切割的腙键或者可以被活性氧类物质切割的氨基丙烯酸酯(AA)接头。
在一个实施方案中,亲水性部分包括亲水性肽、自组装肽、寡核苷酸、水溶性聚合物或经带电侧基官能化的烷基链。优选地,所述烷基链具有多于5个碳原子并且所述带电侧基可以是例如胺基、羧基或胍基(guanidinium group)。
在一个实施方案中,亲水性部分是亲水性肽。例如,亲水性肽可包含含有至少一个Lys、Asp、Arg、His或Glu的氨基酸残基序列。优选的亲水性肽可以是Asp-Asp-Asp-Asp-Asp(SEQ ID NO:1)(“D5”)或Asp-Glu-Val-Asp(SEQ ID NO:2)(“DEVD”)。
优选的自组装肽是(Ala-Glu-Ala-Glu-Ala-Lys-Ala-Lys)2(SEQ ID NO:3)或Phe-Phe。
靶标识别基序优选地对细胞膜受体具有亲和力。优选地,靶标识别基序具有对整联蛋白αvβ3具有亲和力的环(Arg-Gly-Asp)残基(“cRGD”)。或者,所述靶标识别基序可以是溶酶体蛋白跨膜4β。
本发明的发光探针中的发光团至少包括本文中所述的那些荧光团,以及四苯基噻咯(“TPS”)、四苯基乙烯吡啶(“tetraphenylethene pyridinium,PyTPE”)和四苯基乙烯(“TPE”)。
发光探针组合物的组分以线性阵列共价连接。组分以及连接顺序的确定可基于发光探针的期望应用而变化和选择。下表示出了示例性的组分选择以及所述组分的示例性连接顺序(从左向右阅读)。
该表仅用于举例说明目的,并且既不反映所有可能的组分选择也不反映所有可能的连接顺序。例如,当使用AIE-ESIPT荧光团作为发光团时,可移除亲水性部分;当存在两个发光团时,可将第二发光团插入在第三组分(亲水性部分或连接部分)之后但在第四组分(靶标识别基序)之前。
在本发明的发光探针方面的另一个实施方案中,存在具有双重成像功能的特异性AIE发光探针。这样的探针的一个实例是TPE-DEVD-DOTA/Gd(“DDT-Gd”)。
数种示例性探针(例如,表第A、O和X行的探针以及DDT-Gd探针)的详细合成途径以及测试参数和结果可见于本申请的实施例部分。
本发明的发光探针可一般性地用于进行荧光成像和磁共振成像,并且特别地用于评估前药转变成其活性形式、评估前药的治疗效力、检测生物样品中的谷胱甘肽和检测样品中的碱性磷酸酶。下面是对本发明的应用方面的更详细描述。
应用
尽管本发明的发光探针一般性地可用于进行荧光成像和磁共振成像,但是以下讨论将集中于用于治疗响应的非侵入性早期原位评价、药物活化的选择性且实时的原位监测、靶向细胞内硫醇成像和碱性磷酸酶(alkaline phosphatase,ALP)检测的用途。
治疗响应的非侵入性早期原位评价
本发明的发明人已开发了基于本发明的发光探针来实时监测体外和体内细胞凋亡的策略,所述发光探针包含具有AIE(或AIE-ESIPT)特征的荧光团。
因此,本发明该方面的一个实施方案涉及用于评估前药的治疗效力的方法,其包括:
a)将含有活靶细胞的生物样品与本发明的化学组合物在足以将前药转变成其活性形式的条件下孵育以形成经孵育混合物;以及
b)通过荧光光谱术来分析步骤a)的经孵育混合物,
其中与化学组合物不在生物样品存在下的荧光强度相比荧光强度的增加指示活性药物的效力。
特别地参照发光探针TPS-DEVD-Pt-cRGD(参见表格的行A),将如下更详细地解释和说明本发明用于评估前药的治疗效力的方法。
随着对原位监测药物诱导的凋亡的特别关注开发了可靶向的治疗诊断性Pt(IV)前药。治疗诊断性系统(theranostic system)包含在细胞内可被还原成活性Pt(II)的化学治疗性Pt(IV)前药、基于具有AIE特征之三苯基噻咯(TPS)的凋亡传感物(TPS-DEVD)和作为靶向配体的环(RGD)肽(方案1)。前药可优先地在αvβ3整联蛋白过表达的癌细胞中聚集并且在Pt(IV)前药的细胞内还原之后释放活性药物Pt(II)和凋亡传感物TPS-DEVD。释放的Pt(II)可诱导细胞凋亡并且活化胱天蛋白酶-3以切割TPS-DEVD中的DEVD肽并触发荧光。可在用于特定抗癌药物之治疗响应的实时且非侵入性成像的治疗诊断系统中利用荧光开启响应。治疗诊断系统的癌细胞包括例如U87-MG、MDA-MB-231和HT29。所考虑的抗癌药物包括例如多柔比星和紫杉醇。
方案1:具有用于非侵入性原位早期评价其治疗响应的内置聚集诱导发射(AIE)发光凋亡传感物的靶向治疗诊断性铂(IV)前药(TPS-DEVD-Pt-cRGD)的示意图。
药物活化的选择性且实时的原位监测
本发明的发明人利用本发明的发光探针开发了原位检测药物活化的简单策略,所述发光探针包含具有AIE(或AIE-ESIPT)特征的荧光团。
因此,本发明该方面的一个实施方案涉及用于评估前药转变成其活性形式的方法,所述方法包括:
a)将生物样品与上述化学组合物在足以形成经孵育混合物的条件下孵育;以及
b)使用微孔板读取仪来分析步骤a)的经孵育混合物的荧光,
其中与上述化学组合物不在生物样品存在下的荧光强度相比荧光强度的增加指示前药转变成其活性形式。
优选地,该方法在活细胞中进行。
特别地参照发光探针PyTPE-Pt-D5-cRGD(参见表格的行O),下文将更详细地解释和说明本发明用于评估前药转变成其活性形式的方法。
开发了靶向治疗诊断性铂(IV)前药递送系统的设计和合成。该系统基于AIE荧光团来原位监测铂(IV)前药活化。该治疗诊断系统包含可在细胞内部被还原成活性Pt(II)的化学治疗前药Pt(IV)、具有AIE特征的四苯基乙烯吡啶(PyTPE)单元、具有5个天冬氨酸(D5)单元以确保其水溶性的短亲水性肽以及作为靶向配体的环(RGD)肽(cRGD)(方案2)。该前药可优先地在过表达αvβ3整联蛋白的癌细胞中聚集并且可用作针对肿瘤细胞的优异导向分子,所述肿瘤细胞例如U87-MG、MDA-MB-231和HT29细胞。在水性介质中,AIE部分由于D5-cRGD的高亲水性而是非荧光的,但是在Pt(IV)络合物被还原释放而两个轴之后,其发射显著增强。荧光增强(“开启”)有助于限制经切割的残基中PyTPE苯基环的分子内旋转,其占据辐射衰变通道。本发明的前药设计提供了高效靶向铂药物递送以及以高信噪比实时监测药物释放和分布的良好机会。
方案2:前药PyTPE-Pt-D5-cRGD设计策略和荧光开启药物活化的监测
靶向细胞内硫醇成像
本发明的发明人利用本发明的发光探针已开发了用于细胞特异性细胞内硫醇(例如谷胱甘肽)成像的策略,所述发光探针包含具有AIE(或AIE-ESIPT)特征的荧光团。
因此,本发明该方面的一个实施方案涉及用于检测生物样品中的谷胱甘肽的方法,所述方法包括:
a)将认为含有谷胱甘肽的生物样品与上述化学组合物在足以形成经孵育混合物的条件下孵育;以及
b)通过荧光光谱术来分析步骤a)的经孵育混合物,
其中与上述化学组合物不在所述生物样品存在下的荧光强度相比荧光强度的增加指示谷胱甘肽的存在。
在检测谷胱甘肽的方法中,经孵育混合物的荧光强度优选地随谷胱甘肽浓度的增加而增加。
特别地参照发光探针TPE-SS-D5-cRGD(参见表格的行X),下文将更详细地解释和描述本发明用于检测谷胱甘肽的方法。
设计靶向整联蛋白αvβ3的发光探针以进行细胞特异性细胞内硫醇成像。该探针包含靶向环RGD(cRGD)肽、具有5个天冬氨酸(Asp,D5)的高水溶性肽、TPE荧光团和硫醇特异性的可切割二硫键接头。cRGD对αvβ3整联蛋白表现出高结合亲和力,αvβ3整联蛋白是用于早期检测和治疗快速生长实体瘤的独特生物标志物,所述肿瘤包括例如U87-MG、MDA-MB-231和HT29癌细胞。该探针是高水溶性的并且在水性介质中是非荧光的。二硫键基团被硫醇切割导致荧光信号输出增强(方案3)。该探针由此可用于实时监测特定肿瘤细胞中的硫醇(谷胱甘肽)水平。
方案3:(A)一般性探针设计策略和(B)通过αvβ3整联蛋白介导的细胞摄取和二硫键切割来诱导荧光“开启”的cRGD靶向细胞内硫醇成像的示意图。(C)探针TPE-SS-D5-cRGD的化学结构。
碱性磷酸酶(ALP)检测
本发明的发明人利用具有AIE-ESIPT特征的发光探针Phos-HC开发了检测碱性磷酸酶的策略。
因此,本发明该方面的一个实施方案涉及用于检测样品中的碱性磷酸酶的方法,其包括:
a)将认为含有碱性磷酸酶的样品与Phos-HC在足以形成经孵育介质的条件下孵育;以及
b)通过荧光光谱术来分析步骤a)的经孵育介质,
其中在约641nm处的荧光信号荧光强度的增加指示碱性磷酸酶的存在。
该方法的样品优选是活细胞。
对本发明用于检测谷胱甘肽的该方法的更详细说明(包括检测方案和光学结果)可见于本申请的实施例部分。
实施例
AIE荧光团
详细合成途径
在0℃下,向(4-氨基苯基)(苯基)甲酮(1.970g,10mmol)在THF(30mL)中的溶液缓慢地添加氢化钠(1.200g,30mmol,3当量,在油中的60%混悬液),将反应保持2小时,然后注入溴乙烷(2.24mL,30mmol,3当量)。将反应混合物缓慢地升温至室温并搅拌过夜。将溶液用二氯甲烷稀释并用NaHCO3水溶液和盐水洗涤。将有机层经硫酸钠干燥。将滤液浓缩并通过硅胶柱色谱法(乙酸乙酯/己烷=1/10)纯化以产生黄色固体(2.302g,91%)。
在0℃下,向(4-氨基苯基)(苯基)甲酮(0.986g,5mmol)在THF(30mL)中的溶液缓慢地添加氢化钠(0.600g,15mmol,3当量,在油中的60%分散体系),将反应保持2小时,然后注入正溴己烷(2.476g,15mmol,3当量)。将反应混合物缓慢地升温至室温并搅拌过夜。将溶液用二氯甲烷稀释并用NaHCO3水溶液和盐水洗涤。将有机层经硫酸钠干燥。将滤液浓缩并通过硅胶柱色谱法(乙酸乙酯/己烷=1/10)纯化以产生黄色固体(1.605g,88%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入锌粉(1.308g,20mmol)和40mL THF。将混合物冷却至-5℃至0℃,并通过注射器缓慢地添加TiCl4(1.09mL,10mmol),使温度保持在10℃以下。将混悬混合物升温至室温并搅拌0.5小时,然后在回流下加热2.5小时。将混合物再次冷却至-5℃至0℃,装入吡啶(0.5mL,6mmol)并搅拌10分钟。缓慢地添加A(506mg,2mmol)+B(570mg,2mmol)在15mL THF中的溶液。在添加之后,将反应混合物在回流下加热过夜。将反应用10%K2CO3水溶液淬灭并用CH2Cl2萃取。收集有机层并浓缩。通过硅胶柱色谱法(EA/DCM=2∶5)纯化粗制物质以得到期望的黄色产物(0.360g,36%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入锌粉(1.308g,20mmol)和40mL THF。将混合物冷却至-5℃至0℃,并通过注射器缓慢地添加TiCl4(1.09mL,10mmol),使温度保持在10℃以下。将混悬混合物升温至室温并搅拌0.5小时,然后在回流下加热2.5小时。将混合物再次冷却至-5℃至0℃,装入吡啶(0.5mL,6mmol)并搅拌10分钟。缓慢地添加A(731mg,2mmol)+B(570mg,2mmol)在15mL THF中的溶液。在添加之后,将反应混合物在回流下加热过夜。将反应用10%K2CO3水溶液淬灭并用CH2Cl2萃取。收集有机层并浓缩。通过硅胶柱色谱法(EA/DCM=2∶5)纯化粗制物质以得到期望的黄色产物(0.360g,29%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入在甲醇(10mL)中的C2-TPE-Py(100mg,0.197mmol)和1,3-丙磺酸内酯(1,3-Propanesultone)(241mg,1.97mmol)。将反应混合物回流24小时,然后在真空下除去溶剂并对残余物进行硅胶柱色谱法(甲醇/DCM=1/3至纯MeOH)以产生黄色固体(91mg,73%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入在甲醇(10mL)中的C6-TPE-Py(62mg,0.1mmol)和1,3-丙磺酸内酯(122mg,1.0mmol)。将反应混合物回流24小时,然后在真空下除去溶剂并对残余物进行硅胶柱色谱法(甲醇/DCM=1/3至纯MeOH)以产生黄色固体(61mg,82%)。
在0℃下,向(4-氨基苯基)(苯基)甲酮(1.970g,10mmol)在THF(30mL)中的溶液缓慢地添加氢化钠(1.200g,30mmol,3当量,在油中的60%分散体系),将反应保持2小时,然后注入碘甲烷(1.87mL,30mmol,3当量)。将反应混合物缓慢地升温至室温并搅拌过夜。将溶液用二氯甲烷稀释并用NaHCO3水溶液和盐水洗涤。将有机层经硫酸钠干燥。将滤液浓缩并通过硅胶柱色谱法(乙酸乙酯/己烷=1/5)纯化以产生黄色固体(2.010g,89%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入锌粉(2.616mL,20mmol)和40mL THF。将混合物冷却至-5℃至0℃,并通过注射器缓慢地添加TiCl4(2.16mL,20mmol),使温度保持在10℃以下。将混悬混合物升温至室温并搅拌0.5小时,然后在回流下加热2.5小时。将混合物再次冷却至-5℃至0℃,装入吡啶(1.0mL,12mmol)并搅拌10分钟。缓慢地添加A(900mg,4mmol)+B(805mg,4mmol)在30mL THF中的溶液。在添加之后,将反应混合物在回流下加热过夜。将反应用10%K2CO3水溶液淬灭并用CH2Cl2萃取。收集有机层并浓缩。通过硅胶柱色谱法(EA/DCM=2∶5)纯化粗制物质以得到期望的黄色产物(0.280g,23%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入A(2.616g,10mmol)、4-乙烯基吡啶(1.25mL,11mmol)、Pd(OAc)2(90mg,4%mmol)、P(邻甲苯基)3(426mg,14%mmol)、Et3N(36mL)、DMF(24mL),加热至110℃持续30小时。然后,用水稀释反应,将水相用CH2Cl2洗涤并用CHCl3萃取。收集所有的有机层,并蒸发溶剂,收集黄色的粗制产物,然后进行重结晶(EA/CHCl3),获得作为黄色粉末的标题产物(2.600g,91%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入锌粉(2.616mL,20mmol)和40mL THF。将混合物冷却至-5℃至0℃,并通过注射器缓慢地添加TiCl4(2.16mL,20mmol),使温度保持在10℃以下。将混悬混合物升温至室温并搅拌0.5小时,然后在回流下加热2.5小时。将混合物再次冷却至-5℃至0℃,装入吡啶(1.0mL,12mmol)并搅拌10分钟。缓慢地添加A(729mg,4mmol)+B(805mg,4mmol)在30mL THF中的溶液。在添加之后,将反应混合物在回流下加热过夜。将反应用10%K2CO3水溶液淬灭并用CH2Cl2萃取。收集有机层并浓缩。通过硅胶柱色谱法(EA/DCM=2∶5)纯化粗制物质以得到期望的黄色产物(0.230g,22%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入在CH3CN(10mL)中的B(1.00g,10当量)、A(100mg,0.33mmol),将反应混合物回流至少36小时。直至原料A耗尽才淬灭反应。蒸发溶剂并进行柱色谱(DCM,CH3OH),获得作为红色固体的盐(120mg,71%)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入在MeOH(10mL)和THF(5mL)的混合物中的B(52mg,2当量)、A(50mg,0.10mmol),将反应混合物回流至少48小时。直至原料A耗尽才淬灭反应。蒸发溶剂并进行柱色谱(DCM,CH3OH),获得作为红色固体的盐(产率未计算)。
在Ar(g)气氛下,向装备有磁力搅拌器的双颈瓶装入锌粉(1.308g,20mmol)和40mL THF。将混合物冷却至-5℃至0℃,并通过注射器缓慢地添加TiCl4(1.09mL,10mmol),使温度保持在10℃以下。将混悬混合物升温至室温并搅拌0.5小时,然后在回流下加热2.5小时。将混合物再次冷却至-5℃至0℃,装入吡啶(0.5mL,6mmol)并搅拌10分钟。缓慢地添加A(870mg,2mmol)+B(680mg,2mmol)在15mL THF中的溶液。在添加之后,将反应混合物在回流下加热过夜。将反应用10%K2CO3水溶液淬灭并用CH2Cl2萃取。收集有机层并浓缩。通过硅胶柱色谱法纯化混合的粗制物质得到黄色混合物。将混合物放置在密封管中,然后装入4-乙烯基吡啶(0.62mL,5.5mmol)、Pd(OAc)2(45mg,4%mmol)、P(邻甲苯基)3(213mg,14%mmol)、Et3N(12mL)、DMF(8mL)。将反应加热至110℃持续24小时。然后,用水稀释反应,将水相用CH2Cl2洗涤并用CHCl3萃取。收集有机层并浓缩。通过硅胶柱色谱法(EA/DCM=2∶5)纯化粗制物质以得到期望的黄色产物(0.260g,2个步骤中为16%)。
AIE-ESIPT荧光团
DMA-HC的AIE行为
在向DMA-HC的THF溶液添加水之后,溶液发生荧光红移并且在形成聚集体之后增强。参见图1A至1B。
ALP检测
探针1针对溶液中的ALP给出了独特的光学响应。在ALP存在下,溶液的发射最大值从520nm变化至640nm。参见图2A至2B。
相同的探针还可用于细胞ALP检测。参见图3。
该探针由此证明了基于AIE的发光传感和成像的新策略。
发光探针TPS-DEVD-Pt-cRGD
一般性信息
顺铂、N,N-二异丙基乙胺(DIEA)、N-羟基琥珀酰亚胺(NHS)、1-乙基-3-[3-二甲基氨基丙基]碳二亚胺盐酸盐(EDC)、硫酸铜(II)(CuSO4)、抗坏血酸钠、抗坏血酸、琥珀酸酐、3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,MTT)、无水二甲基亚砜(DMSO)、无水二甲基甲酰胺(DMF)、锂线、萘、4-溴苯、4-溴苄基溴、叠氮化钠、二氯双(三苯基膦)钯(II)、ZnCh·TMEDA、哌嗪-N,N′-双(2-乙磺酸)(PIPES)、二乙基二硫代氨基甲酸盐(diethyldithiocarbamate,DDTC)、牛血清白蛋白(BSA)、溶菌酶、胃蛋白酶、胰蛋白酶以及其他化学品全部购买自Sigma-Aldrich并且原样使用而无需进一步纯化。从Fisher Scientific购买的己烷和四氢呋喃(THF)在临用之前从二苯甲酮羰游基钠蒸馏而得。二氯甲烷(DCM)经氢化钙蒸馏。以四甲基硅烷(TMS)作为内部参照物的氘代溶剂购买自Cambridge Isotope Laboratories Inc。炔官能化的DEVD(Asp-GluVal-Asp-Pra)和胺官能化的cRGD(环(Arg-Gly-Asp-D-Phe-Lys))从GL Biochem Ltd.定制。顺,顺,反-二氨二氯二琥珀酸铂(Diamminedichlorodisuccinatoplatinum)(IV)遵循以下文献方法合成[1]。
Dulbecco改良的必需培养基(DMEM)是National University Medical Institutes(Singapore)的商业产品。Milli-Q水由Milli-Q Plus System(Millipore Corporation,Breford,USA)供应。哌嗪-N,N′-双(2-乙磺酸)(PIPES)缓冲液含有50mM PIPES、100mM NaCl、1mM乙二胺四乙酸(EDTA)、0.1%w/v 3-[(3-(胆酰胺基丙基)二甲基铵基]丙磺酸(3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic)和25%w/v蔗糖(pH=7.2)。重组人胱天蛋白酶-3购买自R&D Systems。胱天蛋白酶-3抑制剂5-[(S)-(+)-2-(甲氧基甲基)吡咯烷基]磺酰基靛红购买自Calbiochem。胎牛血清(FBS)和胰蛋白酶-EDTA溶液购买自Gibco(Lige Technologies,AG,Switzerland)。星形孢菌素(Staurosporine,STS)购买自Biovision。DRAQ5购买自Biostatus。经切割的胱天蛋白酶-3(Asp 175)(5A1E)兔mAb(#9664)购买自Cell Signaling。鼠抗-兔IgG-TR(sc-3917)购买自Santa Cruz。
表征
NMR谱在Bruker ARX 400 NMR光谱仪上测量。化学位移参照残余溶剂以百万分之一(parts per million,ppm)报道(CDCl3=7.26ppm、(CD3)2SO=2.50ppm或四甲基硅烷Si(CH3)4=0ppm)。颗粒尺寸及尺寸分布通过用颗粒尺寸分析仪(90 Plus,Btookhaven Instruments Co.,USA)在室温下以90°的固定角度进行激光光散射(laser light scattering,LLS)来确定。HPLC特征谱和质谱使用Shimadzu IT-TOF来获取。使用0.1%TFA/H2O和0.1%TFA/乙腈作为HPLC实验的洗脱剂。高分辨率质谱(HRMS)在Finnigan MAT TSQ 7000质谱仪上记录。UV-vis吸收谱在Milton Ray Spectronic 3000阵列分光光度计上取得。光致发光(PL)谱在Perkin-Elmcr LS 55分光荧光计上测量。细胞用成像软件(Fluoview FV500)通过共聚焦激光扫描显微镜(CLSM,Zeiss LSM 410,Jena,Germany)来成像。图像通过Image J 1.43 x程序(由NIH开发,http://rsbweb.nih.gov/ij/)进行分析。
4-溴苄基叠氮化物的合成
向装备有磁力搅拌器的烧瓶中添加4-溴苄基溴(7.5g,30mmol)、叠氮化钠(7.8g,120mmol)和40mL DMSO。在于70℃下搅拌12小时之后,将溶液倒入150mL的水中并用DCM萃取。通过硅胶柱色谱法使用己烷作为洗脱剂来纯化粗制产物以得到无色的粘性液体,产率为96%(6.12g)。
1H NMR(CDCl3,400MHz),δ(TMS,ppm):7.47(d,2H),7.15(d,2H),4.26(s,2H).13C NMR(CDCl3,100MHz),δ(TMS,ppm):134.3,131.8,129.6,122.1,53.9.HRMS(MALDI-TOF):m/z 210.9640(M+,计算值210.9745).
1,1-二甲基-2-[4-(叠氮基甲基)苯基]-3,4,5-三苯基噻咯(TPS-CH2N3)的合成
根据我们公开的操作制备二甲基双(苯基乙炔基)硅烷。[2]将锂(0.056g,8mmol)和萘(1.04g,8mmol)在8mL THF中的混合物在氮气下在室温下搅拌3小时以形成深墨绿色LiNaph溶液。然后,在室温下,向LiNaph溶液逐滴添加二甲基双(苯基乙炔基)硅烷(0.52g,2mmol)在5mLTHF中的溶液。在搅拌1小时之后,将混合物冷却至室温,然后用25mLTHF稀释。在添加ZnCl2·TMEDA(2g,8mmol)之后形成黑色的混悬液。再在室温下搅拌1小时,之后添加在25mL THF中包含4-溴苯(0.34g,2.2mmol)、4-溴苄基叠氮化物(0.47g,2.2mmol)和PdCl2(PPh3)2(0.08g,0.1mmol)的溶液。将混合物回流过夜。在冷却至室温之后,添加100mL的1M HCl溶液,并用DCM萃取混合物数次。将有机层合并,并用盐水和水洗涤,然后经硫酸镁干燥。在于减压下蒸发溶剂之后,通过硅胶柱使用己烷作为洗脱剂来纯化残余物。获得作为黄色固体产物,产率为36%(0.34g)。
1H NMR(400MHz,CDCl3),δ(TMS,ppm):7.15-7.06(m,6H),7.02-6.99(m,5H),6.95-6.93(m,4H),6.82-6.79(m,4H),4.23(s,2H),0.48(s,6H).13C NMR(CDCl3,100MHz),δ(TMS,ppm):154.5,153.9,142.1,141.2,140.1,139.8,138.7,132.5,130.0,129.2,128.9,128.0,127.5,126.4,126.3,125.7,54.7,-3.80.HRMS(MALDI-TOF):m/z 469.1959(M+,计算值469.1974)。
N-羟基琥珀酰亚胺活化的铂(IV)络合物的合成
将铂(IV)络合物顺,顺,反-二氨二氯二琥珀酸铂(IV)(32.1mg,0.06mmol)、EDC(23.0mg,0.12mmol)和NHS(13.8mg,0.12mmol)在无水DMF(1mL)中的混合物在室温下搅拌过夜。在此之后,混合物通过HPLC(溶剂A:具有0.1%TFA的水;溶剂B:具有0.1%TFA的CH3CN)纯化,并快速冻干以产生作为白色粉末的期望产物,产率为78%(34.1mg)。
1H NMR(400MHz,DMF-d7),δ(TMS,ppm):6.92-6.68(m,6H),2.94-2.91(m,8H),2.89-2.84(m,4H),2.72-2.68(m,4H).13C NMR(DMF-d7,100MHz),δ(TMS,ppm):178.5,170.6,168.8,30.0,27.1,25.9.IT-TOF-MS:m/z[M+H]+计算值728.026,实测值728.021。
凋亡传感物TPS-DEVD-NH2的“点击”合成
将炔官能化的DEVD(10.2mg,20μmol)和TPS-CH2N3(9.4mg,20μmol)溶解在DMSO/H2O溶液(v/v=1/1;1.0mL)的混合物中。通过顺序添加催化量的CuSO4(9.6mg,6μmol)和抗坏血酸钠(2.4mg,12μmol)来起始“点击”反应。在室温下伴随摇动再继续反应24小时。通过HPLC纯化终产物,并在真空下冻干以产生作为白色粉末的探针,产率为45%(9.4mg)。
1H NMR(DMSO-d6,400MHz):12.24(s,3H),8.49(d,1H),8.32(d,1H),8.05(d,1H),7.92(d,1H),7.86(s,1H),7.22-7.06(m,6H),7.02-6.99(m,5H),6.95-6.88(m,4H),6.82-6.79(m,4H),5.45(s,2H),4.54-4.49(m,1H),4.38(m,2H),4.17-4.13(m,2H),3.10-3.05(m,1H),2.92-2.88(m,1H),2.71-2.65(m,2H),2.26-2.21(m,2H),2.01-1.85(m,3H),0.84-0.74(m,6H),0.43(s,6H);IT-TOF-MS:m/z[M+H]+计算值1040.426,实测值1040.866。
治疗诊断性前药TPS-DEVD-Pt-cRGD的合成
将TPS-DEVD-NH2(9.0mg,8.7mmol)和胺官能化的cRGD(5.2mg,8.7mmol)溶解在具有催化量DIEA(1.0μL)的无水DMSO(1.0mL)中。将混合物在室温下搅拌10分钟。然后,向上述混合物快速添加在DMSO(0.5mL)中的N-羟基琥珀酰亚胺活化的铂(IV)复合物(6.3mg,8.7mmol)。在室温下伴随搅拌再继续反应24小时。通过HPLC纯化终产物,并在真空下冻干以产生作为白色粉末的前药,产率为40%(7.4mg)。
1H NMR(DMSO-d6,400MHz):12.24(s,3H),8.55(d,1H),8.51(d,1H),8.31(d,1H),8.15-8.05(m,6H),7.91(d,1H),7.86(s,1H),7.62(d,2H),7.56(d,1H),7.45(m,1H),7.22-7.06(m,6H),7.02-6.99(m,5H),6.95-6.88(m,4H),6.82-6.79(m,4H),6.60-6.35(m,6H),5.45(s,2H),4.65-4.60(m,1H),4.54-4.48(m,1H),4.40-4.32(m,3H),4.17-4.10(m,3H),4.05-4.02(m,1H),3.95-3.91(m,1H),3.10-3.06(m,4H),2.95-2.87(m,2H),2.85-2.78(m,2H),2.75-2.62(m,6H),2.50-2.45(m,8H),2.27-2.23(m,4H),1.93-1.89(m,4H),1.72(m,3H),1.41-1.38(m,2H),1.37-1.32(m,2H),0.84-0.74(m,6H),0.43(s,6H);ESI-MS:m/z[M+H]+计算值2141.719,
实测值2141.689。
用于酶促测定的一般操作
将TPS-DEVD-Pt-cRGD的DMSO储存液用DMSO和PIPES(v/v=1/199)的混合物稀释至10μM。接下来,将每份探针与抗坏血酸或胱天蛋白酶-3在室温下孵育,并测量荧光强度的改变。PL谱在365nm的激发波长下从420至650nm采集。
细胞培养
U87-MG人成胶质细胞瘤癌细胞、MCF-7乳腺癌细胞和293T正常细胞由美国典型培养物保藏中心(American Type Culture Collection,ATCC)提供。将细胞在包含10%经热灭活的FBS(Invitrogen)、100U/mL青霉素和100μg/mL链霉素(Thermo Scientific)的DMEM(Invitrogen,Carlsbad,CA)中培养并维持在具有5%CO2的37℃加湿培养箱中。在实验之前,将细胞预培养至达到汇合。
共聚焦成像
将U87-MG、MCF-7和293T细胞在37℃下在室(LAB-TEK,Chambered Coverglass System)中培养。在80%汇合之后,将培养基移出并用PBS缓冲液洗涤两次。然后,向室添加在DMSO储存液中的探针以达到5μM的终浓度。在一些实验中,在进行前药孵育之前,将细胞与含有cRGD(50μM)或抑制剂(5μM)的培养基预孵育。在于37℃下进行前药孵育2小时之后,用新鲜的培养基更换培养基,在此之后将细胞用冰冷的PBS洗涤两次,用DRAQ5(Biostatus)根据生产商的标准方案活染色细胞核。对于与活性胱天蛋白酶-3抗体的共定位,首先在室温下将细胞用1x PBS中的3.7%甲醛固定15分钟,再用冷PBS洗涤两次,并用PBS中的0.1%Triton X-100透化10分钟。然后,将细胞用1x PBS中的2%BSA封闭30分钟,并用PBS洗涤两次。随后,将细胞与抗-胱天蛋白酶-3抗体/PBS(v/v=1/99)的混合物在室温下孵育1小时,用PBS缓冲液洗涤一次,然后与PBS中的鼠抗兔IgG-TR(0.8μgmL-1)孵育1小时,随后再次用PBS洗涤。然后,立即通过共聚焦激光扫描显微镜(CLSM,Zeiss LSM 410,Jena,Germany)对细胞成像。通过Image J 1.43 x程序(由NIH开发,http://rsbweb.nih.gov/ij/)来分析图像。
通过荧光微孔板读取仪量化细胞凋亡
将U87-MG和MCF-7细胞以4×104细胞mL-1的密度接种在96孔板(Costar,USA)中。在汇合之后,用含不同浓度TPS-DEVD-Pt-cRGD的新鲜的无PBS DMEM培养基更换培养基。在37℃下经历确定的孵育时间之后,将贴壁细胞用1×PBS缓冲液洗涤两次,之后使用T-CAN微孔板读取仪进行荧光测量。激发和发射波长分别为365和480nm。
前药的细胞毒性
使用3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物(MTT)测定来评估U87-MG和MCF-7癌细胞的代谢活性。将细胞以4×104细胞mL-1的密度接种在96孔板(Costar,USA)中。在孵育24小时之后,用前药浓度不同的探针混悬液更换培养基并在37℃下孵育。在指定的时间间隔之后,将孔用1x PBS缓冲液洗涤两次,并向每个孔中添加含100μL新鲜制备MTT(0.5mg mL-1)溶液的培养基。在于培养箱中在37℃下孵育3小时之后,仔细地移除MTT培养基溶液。然后,向每个孔中添加DMSO(100μL),并轻轻地摇动板以使形成的所有沉淀物都溶解。通过微孔板读取仪(Genios Tecan)来监测在570nm处MTT的吸光度。细胞生存力表示为与探针混悬液孵育之细胞的吸光度相对于仅与培养基孵育之细胞的吸光度的比。
结果讨论
治疗诊断性铂(IV)前药TPS-DEVD-Pt-cRGD的合成和表征
通过用4-溴苯和4-溴苄基叠氮化物对二甲基双(苯基乙炔基)硅烷进行异双官能修饰来合成叠氮化物-官能化的四苯基噻咯(TPS-CH2N3)。TPS-CH2N3以及中间体的详细合成和表征示于实验部分和支持信息中。TPS-CH2N3和炔-官能化的DEVD之间通过“点击反应”使用CuSO4/抗坏血酸钠作为催化剂在DMSO/水(v/v=1/1)中的偶联提供了凋亡传感物TPS-DEVD-NH2,在HPLC纯化之后产率为45%。通过分析型HPLC、NMR和HRMS来充分表征该探针的纯度和身份。对可商购获得的抗癌药物顺铂进行修饰以用作TPS-DEVD-NH2和胺官能化cRGD之间的接头。在第一步中,通过过氧化氢来氧化顺铂以产生顺,顺,反-二氨二氯二羟基铂(IV)络合物。接下来,使Pt(IV)络合物与琥珀酸酐在DMSO中在70℃下反应12小时以产生顺,顺,反-二氨二氯二琥珀酸铂(IV)络合物。随后,通过使用EDC作为偶联试剂使羧酸基团与NHS在无水DMF中反应来获得活化的Pt(IV)络合物。通过HPLC纯化活化的Pt(IV)接头并冻干作为白色粉末,产率为78%。用TPS-DEVD-NH2和胺官能化cRGD在无水DMSO中在N,N-二异丙基乙胺(DIEA)存在下不对称地官能化活化的Pt(IV)接头得到期望产物TPS-DEVD-Pt-cRGD,在HPLC纯化之后产率为40%(方案4)。通过HPLC、NMR和HRMS来充分表征该探针的纯度和身份。
方案4:治疗诊断性TPS-DEVD-Pt-cRGD前药的合成途径
对于任何前药,其必须可在修饰之后容易地转变成其原始形式以恢复其治疗能力。为了评价我们的前药作为药物递送系统的潜力,我们研究了在合成的前药被还原之后所形成的Pt(II)物类的性质。据报道,二乙基二硫代氨基甲酸盐(DDTC)可与Pt(II)络合物反应产生加合物Pt(DDTC)2,但不与稳定的Pt(IV)络合物反应。[3][4]在这项工作中,我们使用HPLC-MS系统来监测在DDTC存在下在用抗坏血酸还原之前和之后Pt(IV)络合物的加合物形成。我们选择抗坏血酸作为还原剂,因为其在细胞中高丰度(1mM),其已证明是还原Pt(IV)的主要物质。[5]如图4中所示,顺铂可与DDTC高效地结合形成Pt(DDTC)2加合物。用492.104的质荷比(m/z)通过IT-TOF进一步确认该络合物。另一方面,只有当在DDTC存在下并且使用抗坏血酸处理前药时才能形成Pt(DDTC)2,证明所释放的Pt实体确实是Pt(II)物质。另外,在还原之后形成m/z为1140.344的凋亡传感物TPS-DEVD-COOH。基于这些结果,我们确定Pt(IV)前药在抗坏血酸存在下可被还原同时产生反应性Pt(II)药物和凋亡传感物。
我们接下来研究了我们的前药的光学特性。获得了TPS-CH2N3在THF中以及TPS-DEVD-Pt-cRGD在DMSO/PIPES(v/v=1/199)缓冲液中的UV-vis吸收谱。二者具有类似的吸收特征谱:在320至440nm范围内具有明显的吸光度,而在AIE荧光团被修饰之后略有蓝移。已知,AIE荧光团在良好溶剂中是非荧光的,但是为固体时或者在差溶剂中作为聚集体时强烈发射。[6][7]由图5A中所示的光致发光(PL)谱可见,TPS-CH2N3在DMSO/PIPE(v/v=1/199)的混合物中显示出强烈荧光,而TPS-DEVD-NH2和TPS-DEVD-Pt-cRGD在相同的介质中几乎是非荧光的,这归因于其在水中的优良溶解性。通过激光光散射(LLS)测量来确定疏水性TPS-CH2N3在DMSO/PIPE(v/v=1/199)缓冲液的混合物中的聚集体形成,其显示平均直径为118nm。由于通常在缓冲液中进行生物传感,因此研究离子强度对前药的发射行为的作用是重要的。通过向PS-DEVD-Pt-cRGD的水溶液(10μM)中添加氯化钠来进行实验。当NaCl的浓度从0增加至960mM时,观察到探针的荧光强度几乎无变化。明显地,离子强度不影响TPS-DEVD-Pt-cRGD及其还原产物的荧光特性。其PL特征谱在常用培养基Dulbecco改良的Eagle培养基(DMEM)中也未改变,并且其还原产物在络合物环境中维持“关闭”状态,因此有较大的潜力充当用于以最小的背景干扰进行药物作用研究的特异性发光凋亡传感物。
首先,我们证明了TPS-DEVD-NH2在DMSO/PIPE(v/v=1/199)中的荧光在添加胱天蛋白酶-3(持续60分钟)之后增加。图5B示出了前药在经抗坏血酸还原之后的光学特性。在TPS-DEVD-Pt-cRGD被还原成TPS-DEVD-COOH之后,荧光未改变。为了研究TPS-DEVD-COOH的酶促响应,我们用重组人胱天蛋白酶-3进行了体外酶促测定。制备经抗坏血酸预处理的前药(10μM)和胱天蛋白酶-3(200pM)的混合物,并在PIPES缓冲液中孵育。孵育1小时之后,在425nm至650nm的范围内测量PL谱。如图5B中所示,记录到经抗坏血酸预处理的TPS-DEVD-Pt-cRGD在用胱天蛋白酶-3处理之后的强荧光信号。然而,大部分的荧光通过用胱天蛋白酶-3的高特异性抑制剂5-[(S)-(+)-2-(甲氧基甲基)吡咯烷基]磺酰基靛红预处理探针而容易地竞争掉,[8]表明DEVD从TPS-DEVD-COOH的特异性切割受到抑制。通过LC-MS进一步确认这种胱天蛋白酶-3催化的水解,其中形成m/z为582.659的TPS残余物。在用胱天蛋白酶-3处理之后,TPS残余物形成平均直径为109nm的颗粒,这解释了溶液荧光“开启”。
还在DMSO/PIPE(v/v=1/199)缓冲液的混合物中监测TPS-DEVD-Pt-cRGD溶液(10μM)在添加抗坏血酸和胱天蛋白酶-3之后随时间的荧光改变。如图5C中所示,在与胱天蛋白酶-3孵育之后,观察到溶液中的荧光快速增加。在60分钟内荧光达到平台期(plateau),这是其固有发射的28倍高(图5D)。我们进一步研究了胱天蛋白酶-3浓度对溶液发射的作用。将不同浓度的胱天蛋白酶-3(0至200pM)与TPS-DEVD-Pt-cRGD(10μM)在DMSO/PIPE(v/v=1/199)缓冲液的混合物中孵育1小时,对应谱示于图5E中。随着胱天蛋白酶-3的浓度增加,由于水性介质中形成的TPS聚集体的量增加,PL强度逐渐地增强。如图5F中所示,485nm处的PL强度相对于胱天蛋白酶-3浓度的绘图得到一条完美直线(R2=0.99),表明基于PL强度变化来量化胱天蛋白酶-3的可能性。基于三倍σ方法,胱天蛋白酶-3的检测极限估计是1pM。
为了研究前药的选择性,我们将经抗坏血酸预处理的TPS-DEVD-Pt-cRGD(10μM)与几种蛋白质在相同条件下孵育,所述蛋白质包括溶菌酶、胃蛋白酶、牛血清白蛋白(BSA)和胰蛋白酶。如图6A中所示,仅胱天蛋白酶-3展现出28倍荧光增加,而其他蛋白质的强度仍较低,确认胱天蛋白酶-3特异性地识别并且切割DEVD。该结果证明,我们的探针可用作细胞内部胱天蛋白酶-3的特异性指示物。由于细胞内存在很多不同种类的蛋白质或酶,我们进一步获得了正常和凋亡U87-MG细胞的细胞裂解液,所述凋亡U87-MG细胞被常用的细胞凋亡诱导剂星形孢菌素(STS,2μM)预处理以激活细胞内的胱天蛋白酶-3。[9]将细胞裂解液直接与TPS-DEVD-Pt-cRGD(5μM)孵育,并随时间监测485nm处的荧光强度。如图6B中所示,荧光强度以与图5C中具有胱天蛋白酶-3的溶液研究类似的方式快速增加。与此同时,在与正常细胞的裂解液孵育之后485nm处的荧光强度显示出最小改变,表明前药在经除胱天蛋白酶-3之外的细胞蛋白质处理之后高度稳定。
为了探索使用TPS-DEVD-Pt-cRGD作为靶向药物递送系统以及作为癌细胞中的药物诱导凋亡成像探针的能力,我们首先将TPS-DEVD-NH2与U87-MG细胞在37℃下孵育。在孵育2小时之后,用均可激活细胞凋亡的顺铂或星形孢菌素处理细胞,并用共聚焦显微术进行监测。观察到,正常的未经诱导细胞显示出非常低的荧光强度,表明几乎没有或没有胱天蛋白酶-3活性。形成鲜明对比的是,从经顺铂或形孢菌素处理的细胞采集到强荧光信号。这些结果证明TPS-DEVD-NH2可用作细胞凋亡的指示物。我们接下来将前药TPS-DEVD-Pt-cRGD与U87-MG人成胶质细胞瘤、MCF-7乳腺癌细胞系和正常细胞系293T细胞孵育,共聚焦成像结果示于图7中。选择细胞膜上过表达整联蛋白αvβ3的U87-MG细胞作为整联蛋白阳性癌细胞,而使用具有低整联蛋白αvβ3表达的MCF-7和293T细胞作为阴性对照。在与TPS-DEVD-Pt-cRGD孵育之后,进行所有细胞的实时成像实验。随着孵育时间流逝,U87-MG的荧光随着细胞凋亡进程而逐渐增加,其在6小时时达到最大值。相反,MCF-7和293T细胞甚至在孵育6小时之后仍仅可发现弱荧光信号。当在TPS-DEVD-Pt-cRGD孵育之前用游离的cRGD或/和胱天蛋白酶-3抑制剂预处理U87-MG细胞时,图像显示出弱荧光。这些结果清楚地证明,TPS-DEVD-Pt-cRGD不仅可用于靶向药物递送,而且还具有实时原位检测胱天蛋白酶-3活性的潜力。此外,在探针的共聚焦图像与由抗胱天蛋白酶-3一抗和经Texas Red标记的二抗产生的免疫荧光信号之间观察到优异的重叠(图8)。
我们接下来使用U87-MG和MCF-7细胞二者作为实例比较了前药的细胞中凋亡诱导的荧光强度变化与细胞毒性特征谱之间的关系。在将细胞孵育6小时之后,通过在365nm处激发后监测480nm下的荧光强度。在孵育72小时之后,通过标准的MTT方法来评价细胞的细胞毒性。在该实验中,我们使用U87-MG和MCF-7细胞二者来研究该影响。如图9A至9B中所示,前药对U87-MG细胞具有明显得多的细胞毒性,这应该是由于在其表面上的较高αvβ3整联蛋白表达。凋亡传感物TPS-DEVD-NH2对两种细胞均未显示出显著的细胞毒性。此外,我们可发现,细胞生存力较高的细胞会显示出较低的荧光强度,这意味着细胞凋亡的程度低。例如,当用5μM TPS-DEVD-Pt-cRGD处理两种细胞时,U87-MG细胞在孵育72小时之后细胞生存力仅为31%,而形成鲜明对比的是,MCF-7细胞在相同条件下的细胞生存力为92%。与此同时,关于荧光研究,U87-MG细胞显示出低程度的荧光强度。该结果还与上述凋亡成像非常一致,表明我们的前药确实可充当靶向药物递送载体,以及用于其治疗应答的非侵入性早期原位评价。
结论
总而言之,我们报道了治疗诊断性Pt(IV)前药的合成和生物应用,所述前药用于靶向药物递送以及其治疗应答的早期原位评价。该前药可在细胞内部被还原成活性Pt(II)并且同时在其轴位置释放细胞凋亡传感物。还原的Pt(II)可诱导癌细胞的凋亡并且活化胱天蛋白酶-3。活化的胱天蛋白酶-3进一步切割凋亡传感物的DEVD序列,并且触发TPS残基的AIE效应,由此能够实现以高信噪比早期评价其在细胞中的治疗应答。另外,我们发现当与我们的前药孵育6小时时由凋亡诱导的荧光强度显示与通过MTT测定确定的细胞之细胞生存力(72小时)具有良好的相关性,即,较低的荧光强度将指示较高的细胞生存力,反之亦然。这些结果指示具有内置传感物的治疗诊断性药物递送系统允许快速地评价药物治疗响应,这对于指导治疗决策,例如治疗是否工作良好或是否应停止治疗方案是必需的。
发光探针PyTPE-Pt-D5-cRGD
方案5:PyTPE-Pt-D5-cRGD的合成途径
通过在甲醇中还原叠氮化物-PyTPE(PyTPE-N3)来合成胺官能化的PyTPE(PyTPE-NH2)。五氟苯酚活化的Pt(IV)络合物由可商购获得的抗癌药物顺铂制备而成,并且用作接头。前药PyTPE-Pt-D5-cRGD的合成途径示于方案5中。用PyTPE-NH2和胺官能化的肽D5-cRGD在N,N-二异丙基乙胺存在下不对称地官能化活化的Pt(IV)络合物得到前药,产率为42%。还合成了具有类似结构但不具有cRGD部分的对照前药PyTPE-Pt-D5,产率为44%。另外,通过在偶联反应中使用辛二酸二琥珀酰亚胺酯(disuccinimidyl suberate)替代活化的Pt(IV)络合物制备了不可活化的对照PyTPE-C6-D5-cRGD,产率为46%。NMR和MS表征以高纯度确认了化合物的正确结构。
为了评价我们的前药作为潜在的抗癌药物,我们研究了在还原之后所形成的Pt(II)物质的性质。据报道,二乙基二硫代氨基甲酸盐(DDTC)可与Pt(II)络合物反应产生加合物Pt(DDTC)2,但不与稳定的Pt(IV)络合物反应。[10]使用HPLC-Mass系统来监测Pt(IV)络合物与DDTC在被抗坏血酸还原之后的加合物形成。我们选择抗坏血酸作为还原剂,因为其在细胞内部高丰度,其已证明是还原Pt(IV)的主要化合物。[11]如图10A中所示,顺铂可与DDTC高效地结合形成Pt(DDTC)2加合物。与质荷比(m/z)为492.104的游离DDTC相比,所述加合物在HPLC谱中显示出不同的洗脱时间。此外,只有在抗坏血酸存在下,该前药才可与DDTC反应形成Pt(DDTC)2,确认所释放的Pt实体确实是Pt(II)物质。我们还发现PyTPE-COOH在还原之后的峰显示m/z为1140.344。基于这些结果,我们确定该前药可被抗坏血酸还原产生反应性铂(II)药物并且同时释放轴部分。
获得了PyTPE-NH2在THF中以及PyTPE-Pt-D5-cRGD在DMSO/PBS(磷酸缓冲盐水)混合物(v/v=1/199)中的UV-vis吸收谱。二者在348至500nm范围内具有类似的吸收特征谱,其中最大值在405nm。PyTPE-NH2和PyTPE-Pt-D5-cRGD在DMSO/PBS(v/v=1/199)中的光致发光(PL)谱示于图10B中。疏水性PyTPE-NH2显示出强烈荧光,而PyTPE-Pt-D5-cRGD在相同的混合物中几乎是非荧光的,这是因为TPE苯基环在水性介质中易于分子内旋转。PyTPE-NH2和PyTPE-Pt-D5-cRGD在PL强度方面的显著差异提供了将该前药系统用于实时监测药物活化的机会。
为了研究前药在还原之后的响应,我们将PyTPE-Pt-D5-cRGD(10μM)与抗坏血酸(1mM)在DMSO/PBS(v/v=1/199)中孵育,并在不同时间点测量荧光谱。如图10C中所示,PyTPE-Pt-D5-cRGD的发射强度随时间显著增加,在1.5小时内达到平台期,这是其固有发射的18倍高。非可靶向的前药PyTPE-Pt-D5在孵育之后显示出类似的荧光强度增强。相比之下,对PyTPE-C6-D5-cRGD观察到可忽略的荧光强度的增加。进一步用其他生物酸和蛋白质滴定PyTPE-Pt-D5-cRGD显示出可忽略的荧光强度改变,表明该前药的高稳定性。仅在还原剂(谷胱甘肽和抗坏血酸)存在下,该前药才显示出强烈的荧光改变。这些结果表明荧光增强是由于前药的还原。
接下来,我们将不同浓度的前药与抗坏血酸(1mM)孵育,并监测前药的荧光强度。605nm处的PL强度相对于前药浓度的绘图得到一条完美直线(图10D),表明量化药物活化的可能性。荧光强度逐渐增强归因于水性介质中形成的PyTPE聚集体的量增加。通过激光光散射(LLS)测量来确认探针的分子分解以及经切割产物的聚集体形成。在水性混合物中,从PyTPE-Pt-D5-cRGD溶液未检测到LLS信号。然而,在还原之后,残余的疏水性AIE发光团趋向于聚簇成平均尺寸为145nm的聚集体。非可靶向的探针PyTPE-Pt-D5在与抗坏血酸孵育之后显示出类似的荧光强度的增加。因此,可基于PL强度改变来容易地检测药物活化过程。
将乳腺癌细胞MDA-MB-231的细胞裂解液直接与PyTPE-Pt-D5-cRGD(10μM)孵育,并随时间监测605nm处的荧光强度。荧光强度以与图11的图像C中用抗坏血酸的溶液研究类似的方式快速增加。与此同时,在将PyTPE-Pt-D5-cRGD与裂解液孵育之后,荧光强度显示出最小变化,表明其是高度稳定的对抗性(encounting)细胞蛋白质。
为了探索使用PyTPE-Pt-D5-cRGD监测癌细胞中的靶向细胞内药物还原的能力,将该前药与DA-MB-231和MCF-7乳腺癌细胞系孵育。共聚焦成像结果示于图11中。选择细胞膜上过表达整联蛋白αvβ3的MDA-MB-231细胞作为整联蛋白阳性癌细胞,而使用具有低水平整联蛋白αvβ3表达的MCF-7细胞作为阴性对照。在与PyTPE-Pt-D5-cRGD孵育之后,MDA-MB-231细胞中的荧光随时间逐渐增加,而MCF-7细胞甚至在孵育6小时之后仍仅可发现弱荧光信号(图11的图像D)。相比之下,对于两种细胞系,PyTPE-Pt-D5均以基本相同的行为展现出弱荧光强度(图11和图像B和E)。当在PyTPE-Pt-D5-cRGD孵育之前用游离的cRGD预处理MDA-MB-231细胞时,图像显示出弱荧光。显著差异揭示MDA-MB-231细胞对PyTPE-Pt-D5-cRGD的选择性摄取归因于整联蛋白受体介导的过程。对于PyTPE-C6-D5-cRGD,在孵育6小时之后未观察到可检测的荧光(图11的图像C和F)。
接下来,我们通过MTT测定研究了前药对MDA-MB-231和MCF-7细胞的细胞毒性特征谱。如图12A至12B中所示,PyTPE-Pt-D5-cRGD对MDA-MB-231细胞具有明显得多的细胞毒性,这应该是由于其较高的αvβ3整联蛋白表达。然而,在对MCF-7细胞的平行实验中,其显示出最低细胞毒性。另外,PyTPE-Pt-D5和PyTPE-C6-D5-cRGD对两种细胞均未显示出显著细胞毒性。由这些结果清楚可见,PyTPE-Pt-DS-cRGD中的靶标部分充当针对肿瘤细胞的靶向单元,并且可被还原成毒性Pt(II)物质。
综上所述,我们报道了基于AIE发光团的荧光发光前药的合成和生物应用,所述前药用于实时地检测细胞内的药物活化。由于AIE发光团的独特性质,该前药在水性介质中是非荧光的,但是当在细胞内部被还原之后变得具有高发射性。使用MDA-MB-231作为一个实例,cRGD官能化的肽允许选择性地靶向很多血管源性癌上的αvβ3整联蛋白,这开启了特异性药物递送的新机会。因此,该前药设计开启了特异性肿瘤靶向的新途径,并且其允许通过荧光信号传导改变来检测经活化药物的浓度。
发光探针TPE-SS-D5-cRGD
方案6:TPE-SS-D5-cRGD的合成途径
通过在甲醇中还原叠氮化物-TPE(TPE-CH2N3)来合成胺官能化的TPE(TPE-CH2NH2)。用TPE-CH2NH2和NH2封端的D5-cRGD在无水二甲基亚砜(DMSO)中在N,N-二异丙基乙胺(DIEA)存在下不对称地官能化DSP接头得到探针TPE-SS-D5-cRGD,产率为45%(方案6)。还合成了具有类似结构但不具有cRGD部分的对照探针TPE-SS-D5,产率为49%。另外,通过在偶联反应中使用辛二酸二琥珀酰亚胺酯替代DSP制备了非可活化的对照探针PyTPE-C6-D5,产率为44%。NMR和MS表征以高纯度确认了三种探针的正确结构。
TPE-CH2NH2和TPE-SS-D5-cRGD在DMSO和磷酸缓冲盐水(PBS,pH=7.4)混合物(v/v=1/199)中的光致发光(PL)谱示于图13A。通过使用硫酸喹啉作为标准物,疏水性TPE-CH2NH2作为纳米聚集体显示出量子产率(Φ)为0.23±0.01的强烈荧光。[12]TPE-SS-D5-cRGD探针在相同介质中几乎是非荧光的,(Φ=0.001),这是因为TPE苯基环在水性介质中易于分子内旋转。TPE-CH2NH2和TPE-SS-D5-cRGD在PL强度的显著差异提供了将该探针系统用于硫醇的特异性发光成像的机会。TPE-SS-D5-cRGD的PL谱显示对0至960mM浓度范围的NaCl无响应。其PL特征谱在常用的细胞培养基Dulbecco改良的Eagle培养基(DMEM)中也不改变。该探针在络合物环境中维持“关闭”状态,因此有较大的潜力充当具有最小背景干扰的特异性发光探针。
为了研究该探针对游离硫醇的响应,选择GSH作为代表性硫醇,因为其在人细胞系统中的高浓度。[13]将GSH(1mM)用于与10μMTPE-SS-D5-cRGD在DMSO/PBS混合物(v/v=1/199)中孵育,在不同时间点测量荧光谱。如图13B中所示,TPE-SS-D5-cRGD的发射强度随时间显著增加,在3小时内达到最大值,这是该探针的固有发射的68倍高。TPE-SS-D5在与GSH孵育之后显示出类似的时间依赖性荧光增强,而对TPE-CC-D5观察到可忽略的信号。进一步证明TPE-SS-D5-cRGD在酸性条件下对GSH作出响应。
接下来,我们研究了GSH浓度对探针发射的影响。将不同浓度的GSH(3.9μM至1.0mM)与TPE-SS-D5-cRGD孵育3小时,对应谱示于图13C中。随着GSH浓度增加,由于水性介质中形成的TPE聚集体的量增加,荧光逐渐增强。通过激光光散射(LLS)来确认探针的分子分解以及经切割产物的聚集体形成。在水性混合物中,从TPE-SS-D5-cRGD溶液未能检测到LLS信号。然而,在与GSH孵育之后,残余的疏水性AIE发光团趋向于聚簇成聚集体。通过AFM进一步确认了聚集体的形成。在相同的实验条件下,对TPE-SS-D5观察到类似的荧光强度的增加,但对TPE-CC-D5则不然。另外,将TPE-SS-D5-cRGD在470nm处的PL强度相对于GSH浓度绘图得到一条完美的直线(图13D),表明以使用该探针进行GSH量化的检测极限可能为1.0μM。
为了监测TPE-SS-D5-cRGD的GSH诱导的荧光活化,使用反相HPLC和MS分析来追踪探针对GSH的暴露。在将TPE-SS-D5-cRGD与GSH孵育3小时之后,对混合物进行HPLC分析。除在10.83分钟洗脱的TPE-SS-D5-cRGD峰之外,还观察到两个新峰:GSS-TPE在10.68分钟和TPE-SH在11.58分钟,并且通过IT-TOF分析,所述峰分别显示出755.217和472.164的质荷比(m/z)。TPE-SH和GSS-TPE的片段在DMSO/PBS(v/v=1/199)中倾向于聚集,使用硫酸喹啉作为参照物,其分别显示出量子产率为19±1%和12±1%的蓝色荧光。这些结果清楚地证明,所观察到的TPE-SS-D5-cRGD之GSH诱导的荧光强度改变归因于二硫键的切割,其导致探针和片段之间的溶解性差异。用GSH中含有的三种氨基酸半胱氨酸(Cys)、甘氨酸(Gly)和谷氨酸(Giu)进一步滴定TPE-SS-D5-cRGD,揭示荧光开启归因于Cys中的游离巯基与二硫键的相互作用。
为了探索使用TPE-SS-D5-cRGD作为用于监测癌细胞中的细胞内硫醇水平的特异性生物探针的能力,将该探针与U87-MG人成胶质细胞瘤和MCF-7乳腺癌细胞系孵育。共聚焦成像结果示于图14中。选择细胞膜上过表达整联蛋白αvβ3的U87-MG细胞作为整联蛋白阳性癌细胞,而使用具有低水平整联蛋白αvβ3表达的乳腺癌细胞MCF-7作为阴性对照。在与TPE-SS-D5-cRGD孵育之后,对U87-MG细胞观察到强蓝色荧光(图14的图像A),而对于MCF-7细胞甚至在孵育6小时之后也仅可发现弱荧光信号(图14的图像D)。相比之下,TPE-SS-D5对于两种细胞系均以基本相同的行为展现出弱荧光强度(图14的图像B和E)。当在TPE-SS-D5-cRGD孵育之前用游离的cRGD预处理U87-MG细胞时,图像显示出弱荧光。显著差异揭示U87-MG细胞对TPE-SS-D5-cRGD的选择性摄取归因于整联蛋白受体介导的过程。对于对照探针TPE-CC-D5,甚至在孵育6小时之后仍未观察到可检测的荧光(图14的图像C和F)。应当注意,该探针还可用于活细胞成像。
为了提供硫醇诱导的二硫键切割作为荧光开启之触发的更多证据,还将U87-MG细胞用丁硫氨酸亚砜亚胺(buthionine sulfoximine,BSO)预处理,之后与TPE-SS-D5-cRGD孵育。BSO是g-谷氨酰半胱氨酸合成酶(g-glutamylcysteine synthetase)的抑制剂,其可抑制细胞合成GSH。[14]经TPE-SS-D5-cRGD处理U87-MG细胞的荧光随BSO的浓度从25μM增加至100μM而降低。与图14的图像A中相比荧光显著降低揭示探针荧光与细胞中的GSH浓度直接相关。这些结果表明,尽管细胞中存在其他的游离硫醇,但是仍可使用TPE-SS-D5-cRGD作为细胞内GSH成像的指示物。体外细胞毒性研究还显示TPE-SS-D5-cRGD探针是生物相容的。
综上所述,我们报道了GSH响应性发光AIE探针的合成和生物应用。由于AIE发光团的独特性质,该探针在水性介质中是非荧光的,但是当被硫醇切割之后变得具有高发射性。该探针能够实现以高信噪比发光监测溶液和细胞中的游离硫醇。使用U87-MG作为一个实例,cRGD官能化的肽允许选择性地靶向很多血管源性癌上的αvβ3整联蛋白,这开启了特异性细胞内硫醇成像的新机会。我们的探针策略可通过简单地用化学生物学中的其他可切割接头改变二硫键基团而推广用于执行多种任务。
发光探针DDT-Gd
TPE-DEVD-DOTA/Gd(DDT-Gd)的合成途径如下:
DEVD-TPE的合成
将过量具有炔的肽(DEVD-炔,22mg,38.7μmol)和叠氮化物官能化的TPE(TPE-CH2N3,10mg,25.8μmol)溶解在0.8mL DMSO中,并涡旋以获得澄清溶液。随后,向混合物中添加溶解于0.2mL Milli-Q水中的CuSO4(0.5mg,9.6μmol)和抗坏血酸钠(2.5mg,38.7μmol)以起始点击化学。使反应在37℃下于摇动下进行约2天。然后,通过HPLC纯化产物TPE-DEVD(产率为60%),并通过LC-MS和NMR进一步表征。
DOTA-DEVD-TPE(DDT)的合成
将这样合成的DEVD-TPE(10mg,10.4μmol)和过量的DOTA-NHS酯(10.4mg,20.8μmol)溶解在总共0.6mL DMSO中,并通过涡旋彻底混合。允许反应在室温下于摇动下进行约2天。然后,通过HPLC纯化产物DDT(产率为70%),并通过LC-MS和NMR进一步表征。IT-TOF-MS:m/z[M+2H]2+计算值672.799,实测值672.782。
DDT-Gd的合成
将这样合成的DDT产物(10mg,7.4μmol)溶解在0.4mL DMSO中。将GdCl3(9.8mg,37μmol)溶解在0.4mL pH使用NaOH调节至5的Milli-Q水中。然后,将GdCl3溶液添加到DDT中,并将混合物通过涡旋彻底混合。在室温下摇动反应混合物以进一步反应约4天。然后,通过HPLC纯化产物DDT-Gd(产率为60%),并通过LC-MS和NMR进一步表征。IT-TOF-MS:m/z[M+2H]2+计算值750.249,实测值750.222。
参考文献
[1]K.R.Barnes,A.Kutikov,S.J.Lippard,Chemistry&Biology 2004,11,557.
[2]J.Z.Liu,R.H.Zheng,Y.H.Tang,M.Haussler,J.W.Y.Lam,A.Qin,M.X.Ye,Y.N.Hong,P.Gao,B.Z.Tang,Macromolecules 2007,40,7473.
[3]A.Lopez-Flores,R.Jurado,P.Garcia-Lopez,J Pharmacal Toxicol Methods 2005,52,366.
[4]J.Li,S.Q.Yap,C.F.Chin,Q.Tian,S.L.Yoong,G.Pastorin,W.H.Ang,Chemical Science 2012,3,2083.
[5]N.Graf,S.J.Lippard,Advanced Drug Delivery Reviews 2012,64,993.
[6]K.L.D.Ding,B.Liu,and B.Z.Tang,Acc.Chem.Res.2013,DO1:10.1021/ar3003464.
[7]Y.N.Hong,J.W.Y.Lam,B.Z.Tang,Chemical Society Reviews 2011,40,5361.
[8]D.Lee,S.A.Long,J.L.Adams,G.Chan,K.S.Vaidya,T.A.Francis,K.Kikly,J.D.Winkler,C.M.Sung,C.Debouek,S.Richardson,M.A.Levy,W.E.DeWolf,P.M.Keller,T.Tomaszek,M.S.Head,M.D.Ryan,R.C.Haltiwanger,P.H.Liang,C.A.Janson,P.J.McDevitt,K.Johanson,N.0.Concha,W.Chan,S.S.Abdel-Meguid,A.M.Badger,M.W.Lark,D.P.Nadeau,L.J.Suva,M.Gowen,M.E.Nuttall,Journal of Biological Chemistry 2000,275,16007.
[9]A.Luhrmann,C.V.Nogueira,K.L.Carey,C.R.Roy,Proceedings of the National Academy of Sciences of the United States of America 2010,107,18997.
[10]J.Li,S.Q.Yap,C.F.Chin,Q.Tian,S.L.Yoong,G.Pastorin and W.H.Ang,Chemical Science,2012,3,2083-2087.
[11]N.Graf and S.J.Lippard,Advanced Drug Delivery Reviews,2012,64,993-1004.
[12]Demas,J.N.;Crosby,G.A.J.Phys.Chem.1971,75,991.
[13]Hwang,C.;Sinskey,A.J.;Lodish,H.F.Science 1992,257,1496.
[14]Hultberg,M.;Hultberg,B.Chem.Biol. Interact.2006,163,192.
本文中引用的所有专利、公开申请和参考文献的教导均通过引用以其整体并入。
尽管已经参照本发明的示例性实施方案对本发明进行了具体示出和描述,但是本领域技术人员将了解,可在本发明中进行形式和细节的多种改变而不偏离所附权利要求书涵盖的本发明的范围。
- 还没有人留言评论。精彩留言会获得点赞!