一种桂枝茯苓胶囊中多糖含量的检测方法与流程
本发明属于中药检测领域,尤其涉及一种桂枝茯苓胶囊中多糖含量的检测方法。
背景技术:
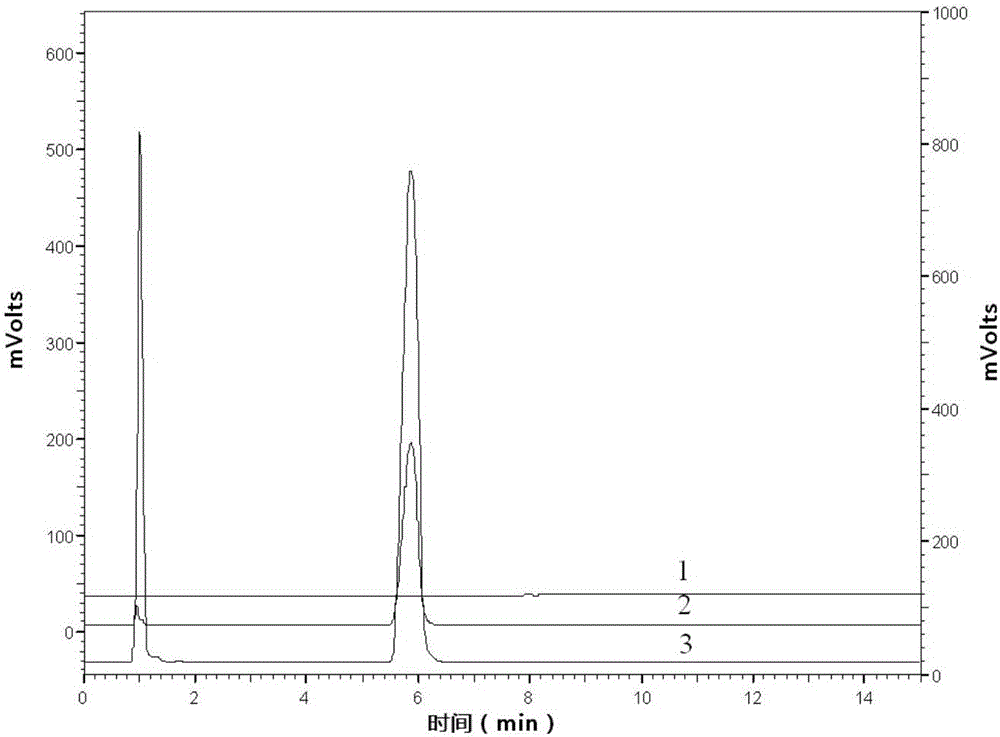
:桂枝茯苓胶囊由桂枝、茯苓、牡丹皮、赤芍和桃仁共五味中药组成,具有活血、化瘀、消癥之功效,主治妇人瘀血阻络所致癥块、经闭、痛经、产后恶露不尽;子宫肌瘤,慢性盆腔炎包块,痛经,子宫内膜异位症,卵巢囊肿见上述证候者;也可用于女性乳腺香囊性增生病属瘀血阻络症,症见乳房疼痛、乳房肿块、胸肋胀闷;或用于前列腺增生属瘀阻膀胱证,症见小便不爽、小腹胀痛。桂枝茯苓胶囊中的多糖含量对控制和评价桂枝茯苓胶囊的质量具有十分重要的意义。目前多糖含量的检测多采用显色法,这种方法专属性差,且准确度不高。因此,为了更好地反映桂枝茯苓胶囊的内在质量,研究一种新的桂枝茯苓胶囊中多糖含量的检测方法是十分必要的。技术实现要素:有鉴于此,本发明的目的在于提供一种桂枝茯苓胶囊中多糖含量的检测方法,本发明提供的方法专属性好,准确性高。本发明提供了一种桂枝茯苓胶囊中多糖含量的检测检测方法,包括以下步骤:a)、将水与桂枝茯苓胶囊的内容物混合后依次进行超声处理和固液分离,得到沉淀物;b)、将所述沉淀物与硫酸混合,回流反应,得到反应液;c)、将所述反应液的pH值调节至4~8后与乙醇混合,得到待测样品液;d)、采用高效液相色谱-蒸发光散射检测器对所述待测样品液进行检测,根据检测结果和预先建立的标准曲线计算,得到所述待测样品液中D-无水葡萄糖的含量。优选的,所述内容物与水的用量比为(0.1~0.5)g:(20~50)mL。优选的,所述硫酸的浓度为2~5mol/mL;优选的,所述内容物与硫酸的用量比为(0.1~0.5)g:(10~30)mL。优选的,所述回流反应的时间为2~6h。优选的,步骤c)中,将所述反应液的pH值调节至4.5~6。优选的,步骤d)中,检测过程中采用的流动相为三乙胺溶液和乙腈。优选的,步骤d)中,检测过程中的流动相流速为0.8~1.2mL/min。优选的,步骤d)中,检测过程中的色谱柱柱温为40~50℃。优选的,步骤d)中,检测过程中的理论塔板数按D-无水葡萄糖峰计算不低于2000。与现有技术相比,本发明提供了一种桂枝茯苓胶囊中多糖含量的检测方法,该方法包括以下步骤:a)、将水与桂枝茯苓胶囊的内容物混合后依次进行超声处理和固液分离,得到沉淀物;b)、将所述沉淀物与硫酸混合,回流反应,得到反应液;c)、将所述反应液的pH值调节至4~8后与乙醇混合,得到待测样品液;d)、采用高效液相色谱-蒸发光散射检测器对所述待测样品液进行检测,根据检测结果和预先建立的标准曲线计算,得到所述待测样品液中D-无水葡萄糖的含量。本发明首先以水为溶剂在超声条件下溶解桂枝茯苓胶囊的内容物中单糖、二糖和β-环糊精;然后固液分离,得到含有多糖成分的沉淀物;之后将该沉淀物与硫酸混合反应,使沉淀物中的多糖水解为D-无水葡萄糖;最后将反应得到的反应液制成待测样品液,采用HPLC(高效液相色谱)-ELSD(蒸发光散射)法对待测样品液中的D-无水葡萄糖的含量进行检测。由于沉淀物中的糖类成分只有多糖,待测样品液中的D-无水葡萄糖均由多糖水解而成,因此通过测定待测样品液中D-无水葡萄糖含量的大小可直接反映出桂枝茯苓胶囊中多糖含量的高低,从而实现对桂枝茯苓胶囊中多糖含量的评价。对本发明提供的方法的专属性、精密度、稳定性、重复性和回收率进行评价,结果表明本发明提供的方法专属性良好,精密度RSD≤1.16%,稳定性RSD≤2.21%,重复性RSD为1.89%,平均回收率为100.11%、回收率RSD为1.39%,可用于评价桂枝茯苓胶囊中的多糖含量。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。图1是本发明实施例1提供的D-无水葡萄糖的浓度-峰面积标准曲线;图2是本发明实施例5提供的D-无水葡萄糖含量测定专属性试验图谱;图3是本发明实施例6提供的不同流速和柱温的色谱图;图4是本发明实施例6提供的不同载气流速、漂移管温度和色谱柱的色谱图。具体实施方式下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明提供了一种桂枝茯苓胶囊中多糖含量的检测方法,包括以下步骤:a)、将水与桂枝茯苓胶囊的内容物混合后依次进行超声处理和离心,得到沉淀物;b)、将所述沉淀物与硫酸混合,回流反应,得到反应液;c)、将所述反应液的pH值调节至4~8后与乙醇混合,得到待测样品液;d)、采用高效液相色谱-蒸发光散射检测器对所述待测样品液进行检测,根据检测结果和预先建立的标准曲线计算,得到桂枝茯苓胶囊中的多糖含量。在本发明提供的方法中,首先除去桂枝茯苓胶囊内容物中的单糖、二糖和β-环糊精,采用的方式为:a)、将水与桂枝茯苓胶囊的内容物混合后依次进行超声处理和固液分离,得到沉淀物。其中,所述内容物和水的用量比优选为(0.1~0.5)g:(20~50)mL,更优选为0.25g:(20~50)mL,最优选为0.25g:30mL。所述超声处理的频率优选为20~60KHz,更优选为30~40KHz;所述超声处理的功率优选为200~300W,更优选为250~260W;所述超声处理的时间优选为10~60min;更优选为30~40min。所述固液分离的方式优选为离心;所述离心的转速优选为5000~20000r/min,更优选为10000~15000r/min,最优选为11000~13000r/min;所述离心的时间优选为10~30min,更优选为15~20min。在本发明中,为了提高沉淀物中多糖的纯净度(减少单糖、二糖和β-环糊精含量),优选对得到的所述沉淀物重复进行步骤a)的操作,所述重复的次数优选为1~3次,更优选为2次。得到所述沉淀物中,将所述沉淀物与硫酸混合,进行回流反应。其中,所述硫酸的浓度优选为2~5mol/mL,更优选为3mol/mL。所述内容物与硫酸的用量比优选为(0.1~0.5)g:(10~30)mL,更优选为0.25g:(10~30)mL,最优选为0.25g:20mL。所述回流反应的时间优选为2~6h,更优选为3~4h。回流反应过程中桂枝茯苓胶囊中多糖水解为D-无水葡萄糖,回流反应结束后,得到反应液。得到所述反应液后,将所述反应液的pH值调节至4~8,优选调节至4.5~6。在本发明提供的一个实施例中,使用氢氧化钠溶液条件所述反应液的pH值,所述氢氧化钠溶液的浓度优选为35~40wt%。pH调节完毕后,将其与乙醇混合,得到待测样品液。在本发明中,优选先对完成pH调节的反应液进行减压浓缩,之后再将其与乙醇混合,得到待测样品液。在本发明中,优选先将减压浓缩的反应液与部分乙醇混合,之后固液分离,液相再与余量的乙醇混合,得到待测样品液。在本发明提供的一个实施例中,所述待测样品液中的乙醇浓度为70~80vol%。在本发明提供的一个实施例中,所述内容物与所述待测样品液的质量体积比优选为(0.1~0.5)g:(50~200)mL,更优选为0.25g:(50~200)mL,最优选为:0.25g:100mL。得到所述待测样品液后,采用高效液相色谱-蒸发光散射检测器对所述待测样品液进行检测。检测过程中,采用的流动相优选为三乙胺溶液和乙腈;所述三乙胺溶液的浓度优选为0.1~0.2vol%;所述三乙胺溶液和乙腈的体积比优选为(10~30):(90~70),更优选为20:80。检测过程中,流动相流速优选为0.8~1.2mL/min,更优选为1mL/min。检测过程中,色谱柱优选为XBridgeAmide型色谱柱;所述色谱柱的填充剂优选为以十八烷基硅烷键合硅胶;色谱柱的柱温优选为40~50℃,更优选为45℃。检测过程中,漂移管的温度优选为90~100℃,更优选为95℃。检测过程中,载气的气体流速优选为2~3L/min。检测过程中,理论塔板数按D-无水葡萄糖峰计算优选不低于2000。检测结束后,得到待测样品液中D-无水葡萄糖的峰面积,根据所述待测样品液中D-无水葡萄糖的峰面积与预先建立的D-无水葡萄糖的浓度-峰面积标准曲线,得到待测样品液中D-无水葡萄糖的含量。本发明对所述D-无水葡萄糖的浓度-峰面积标准曲线建立方式没有特别限定,可按照以下方式建立:首先,提供一系列D-无水葡萄糖的标准溶液;然后,采用高效液相色谱-蒸发光散射检测器对所述一系列D-无水葡萄糖的标准溶液进行检测,得到D-无水葡萄糖每个浓度的标准溶液的色谱图;最后,根据所述色谱图与其对应的标准溶液的浓度,得到D-无水葡萄糖的浓度-峰面积标准曲线。在本发明提供的上述获得D-无水葡萄糖的浓度-峰面积标准曲线的方法中,首先提供一系列不同浓度的D-无水葡萄糖的标准溶液,所述标准溶液由D-无水葡萄糖与乙醇配制得到,所述乙醇的浓度优选为70~80vol%。在本发明提供的一个实施例中,一系列不同浓度的所述D-无水葡萄糖的标准溶液的浓度为200~2100μg/mL;在本发明提供的另一个实施例中,一系列不同浓度的所述D-无水葡萄糖的标准溶液的浓度分别为201.8μg/mL、504.5μg/mL、807.2μg/mL、1109.9μg/mL、1412.6μg/mL、1715.3μg/mL和2018μg/mL。得到一系列D-无水葡萄糖浓度的标准溶液后,采用高效液相色谱-蒸发光散射检测器对所述一系列D-无水葡萄糖浓度的标准溶液进行检测,得到每个浓度的D-无水葡萄糖标准溶液的色谱图。在本发明中,所述标准溶液的检测色谱条件与所述待测样品液进行检测时的色谱条件一致,在此不再赘述。得到每个浓度的D-无水葡萄糖标准溶液的色谱图后,本发明计算得到每个浓度的D-无水葡萄糖标准溶液的峰面积,根据得到的峰面积与其对应的D-无水葡萄糖标准溶液的浓度,得到D-无水葡萄糖浓度-峰面积标准曲线。由于沉淀物中的糖类成分只有多糖,待测样品液中的D-无水葡萄糖均由多糖水解而成,因此得到待测样品液中D-无水葡萄糖含量后可根据其含量的大小直接反映出桂枝茯苓胶囊中多糖含量的高低,从而实现对桂枝茯苓胶囊中多糖含量的评价。本发明首先以水为溶剂在超声条件下溶解桂枝茯苓胶囊的内容物中单糖、二糖和β-环糊精;然后固液分离,得到含有多糖成分的沉淀物;之后将该沉淀物与硫酸混合反应,使沉淀物中的多糖水解为D-无水葡萄糖;最后将反应得到的反应液制成待测样品液,采用HPLC(高效液相色谱)-ELSD(蒸发光散射检测)法对待测样品液中的D-无水葡萄糖的含量进行检测。由于沉淀物中的糖类成分只有多糖,待测样品液中的D-无水葡萄糖均由多糖水解而成,因此通过测定待测样品液中D-无水葡萄糖含量的大小可直接反映出桂枝茯苓胶囊中多糖含量的高低,从而实现对桂枝茯苓胶囊中多糖含量的评价。采用这种方法评价桂枝茯苓胶囊中多糖含量具有较好的专属性和较高的准确性。对本发明提供的方法的专属性、精密度、稳定性、重复性和回收率进行评价,结果表明本发明提供的方法专属性良好,精密度RSD≤1.16%,稳定性RSD≤2.21%,重复性RSD为1.89%,平均回收率为100.11%、回收率RSD为1.39%,可用于评价桂枝茯苓胶囊中的多糖含量。为更清楚起见,下面通过以下实施例进行详细说明。在本发明中,下述实施例涉及使用以下仪器和试剂:1、仪器:MettlerAE240电子分析天平;SartoriusBP211D电子分析天平;KQ-250DB数控超声清洗仪;HH数显恒温水浴锅;高效液相色谱-ELSD检测器:(1)Agilent1100高效液相色谱仪,ELSD检测器;(2)Agilent1200高效液相色谱仪,ELSD检测器。2、试剂:桂枝茯苓胶囊(批号:140301、140701、140801、140901;江苏康缘药业股份有限公司);D-无水葡萄糖(中国食品药品检定研究院,批号:1110833-201205,供含量测定用);乙腈(色谱纯,上海星可生化有限公司);超纯水(自制);其它试剂均为分析纯。实施例1建立标准曲线1、色谱条件色谱柱:XbridgeAmide(3.5μm,4.6×150mm);以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1vol%三乙胺溶液(体积比=80:20)为流动相;色谱柱柱温45℃;流动相流速1.0mL/min;ELSD:漂移管温度95℃;载气气体流速2.0L/min;理论塔板数以D-无水葡萄糖计算应不低于2000。2、建立D-无水葡萄糖的浓度-峰面积标准曲线取D-无水葡萄糖适量,精密称定,加80vol%乙醇制成201.8μg/mL、504.5μg/mL、807.2μg/mL、1109.9μg/mL、1412.6μg/mL、1715.3μg/mL、2018μg/mL的D-无水葡萄糖标准溶液,分别精密吸取上述溶液各10μL,注入高效液相色谱-ELSD检测器,进行检测,记录色谱峰峰面积。每个浓度的标准溶液检测两次,以进样量(μg/mL)对数为横坐标(X),两次检测的平均峰面积的对数为纵坐标(Y),绘制标准曲线,结果如表1和图1所示。表1是本发明提供的D-无水葡萄糖线性关系考察结果,图1是本发明实施例1提供的D-无水葡萄糖的浓度-峰面积标准曲线。表1D-无水葡萄糖线性关系考察结果根据表1计算回归方程及相关系数为:Y=1.8242X+1.5221;R2=0.9998。可见,D-无水葡萄糖进样量在201.8~2018μg/mL之间呈良好线性关系。结果见表1及图1。实施例2桂枝茯苓胶囊中多糖含量的评价1、待测样品液的制备:取10粒桂枝茯苓胶囊(批号:140301)的内容物,精密称定,研细,取约0.25g,精密称定,置具塞锥形瓶中,加入纯化水30mL。超声处理(40KHz,250W)30分钟,放冷,离心(11000r/min,15min),弃去上清液,沉淀加纯化水重新超声处理30min,离心(11000r/min,15min),将锥形瓶内剩余样品加入30mL纯化水少量多次的转移至离心管内,离心(11000r/min,15min),离心后沉淀加3mol/mL硫酸20mL少量多次转移至回流瓶内,于沸水浴中回流3h,放冷,加35wt%氢氧化钠调节pH值至4.5~6.0,减压浓缩,加乙醇溶解,使溶液乙醇浓度为70~80vol%,放冷,离心,溶液加80vol%乙醇定容至100mL,摇匀,得到桂枝茯苓胶囊的待测样品液。2、D-无水葡萄糖含量的测定取按照上述方法制备的待测样品液6份,分别精密吸取上述待测样品液各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到待测样品液中D-无水葡萄糖的含量,结果如表2所示:表2D-无水葡萄糖的含量计算结果编号1(wt%)2(wt%)3(wt%)4(wt%)5(wt%)6(wt%)平均含量RSD(%)D-无水葡萄糖35.1935.9335.3336.6936.8136.3336.051.89通过表2可以看出,该方法重复性良好,待测样品液中D-无水葡萄糖的平均含量为36.05wt%,RSD为1.89%。实施例3系统适用性试验1、对照品溶液的制备取D-无水葡萄糖对照品适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖0.8mg的溶液,即得对照品溶液。2、供试品溶液的制备:按照实施例2中所述待测样品液的制备方法制得供试品溶液。3、测定精密吸取对照品溶液5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,连续重复进样5次,计算峰面积测量值的相对标准偏差;精密吸取供试品溶液5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,计算待测组分的理论塔板数、分离度与拖尾因子。结果如表3所示:表3桂枝茯苓胶囊系统适应性主要参数结果表在上述条件下,拖尾因子为0.92~0.95,D-无水葡萄糖的色谱系统峰面积重复性相对标准偏差为0.53%,以D-无水葡萄糖峰计算理论板数大于2000,为确保定量分析的准确,规定理论板数按D-无水葡萄糖峰计算不得低于2000。实施例4待测样品液制备过程中条件参数的影响1、水提取次数的影响取10粒桂枝茯苓胶囊(批号:140301)的内容物,精密称定,研细,取约0.25g,精密称定,置具塞锥形瓶中,加入纯化水30mL。超声处理(40KHz,250W)30分钟,放冷,离心(11000r/min,15min),得到上清液(即一次提取液)和沉淀,沉淀加纯化水重新超声处理30min,离心(11000r/min,15min),得到上清液(即二次提取液)和沉淀,将锥形瓶内沉淀加入30mL纯化水少量多次的转移至离心管内,离心(11000r/min,15min),得到上清液(即三次提取液)和沉淀。对一次提取液、二次提取液和三次提取液中的果糖、葡萄糖、蔗糖和β-环糊精含量进行测定,结果如表4所示:表4提取次数考察结果提取次数果糖(wt%)葡萄糖(wt%)蔗糖(wt%)β-环糊精(wt%)第1次6.017.4316.437.68第2次0.570.871.063.16第3次无无无0.14实验结果显示:用水提取三次后基本上将单糖、二糖和环糊精去除干净。2、硫酸浓度的影响取批号为140301的桂枝茯苓胶囊内容物,按照实施例2中待测样品液的制备方法,分别选择2mol/mL、3mol/mL、4mol/mL和5mol/mL的硫酸制备2份待测样品液,标记为①和②。分别精密吸取上述待测样品液各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到待测样品液中D-无水葡萄糖的含量,结果如表5所示:表5不同浓度硫酸对D-无水葡萄糖含量测定的影响通过表5可以看出,不同的硫酸浓度对多糖水解有一定影响,3mol/mL为最佳浓度。3、不同pH值的影响取批号为140301的桂枝茯苓胶囊内容物,按照实施例2中待测样品液的制备方法,在“加35wt%氢氧化钠中和至PH为4.5~6”时,分别调至不同的PH值,通过回收率以考察不同pH值对D-无水葡萄糖含量检测的影响;分别精密吸取上述待测样品液各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到待测样品液中D-无水葡萄糖的含量,进而计算出回收率,结果如表6所示。表6中,“称样量”是指制备供试品溶液时所称取的桂枝茯苓胶囊内容物的量,“含量”是指称取的内容物中理论上含有的D-无水葡萄糖糖的量,“加入量”是指加入的D-无水葡萄糖对照品的量,“实测值”是指供试品溶液中测得的D-无水葡萄糖的量。表6不同pH对回收率的影响编号PH值称样量(mg)含量(mg)加入量(mg)实测值(mg)回收率(%)110.9125.848.9038.1176.4472.2824.11125.248.6738.0680.7384.2534.48125.648.8238.0881.0484.6144.89124.648.4338.1580.9585.2656.02125.648.8238.0280.8984.3565.59126.249.0538.1380.6582.89通过表6中的数据可以看出,强碱条件能破坏多糖的水解。4、不同回流时间的影响取批号为140301的桂枝茯苓胶囊内容物,按照实施例2中待测样品液的制备方法,分别选择2h、3h、4h、5h和6h的回流时间制备待测样品液。分别精密吸取上述待测样品液各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到待测样品液中D-无水葡萄糖的含量,结果如表7所示:表7不同回流时间考察试验结果编号回流时间含量(wt%)12h36.5323h40.4934h39.6645h34.9656h32.93通过表5可以看出,不同的回流时间对多糖水解有一定影响,3h为最佳回流时间。实施例5专属性试验1、对照品溶液的制备:取D-无水葡萄糖对照品适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖0.8mg的溶液,即得对照品溶液。2、供试品溶液的制备:按照实施例2中所述待测样品液的制备方法制得供试品溶液。3、阴性供试品溶液的制备除不加样品外,按供试品溶液制备方法制得阴性供试品溶液。4、测定精密吸取对照品溶液5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,结果如图2所示,图2是本发明实施例5提供的D-无水葡萄糖含量测定专属性试验图谱,图2中1为阴性供试品、2为对照品、3为供试品。通过图2可以看出,本发明提供的方法的专属性良好。实施例6耐用性试验1、不同流动相流速和色谱柱柱温的影响取D-无水葡萄糖对照品适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖1000μg的溶液,即得对照品溶液。精密吸取上述对照品溶液5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件,分别在0.9mL/min、1.0mL/min和1.1mL/min的流动相流速条件下,以及43℃、45℃和47℃的流动相流速条件下进行检测,记录色谱峰峰面积,结果如图3所示,图3是本发明实施例6提供的不同流速和柱温的色谱图,图3中1为流速=1.0mL/min、T=47℃,2为流速=1.0mL/min、T=43℃,3为流速=1.1mL/min、T=45℃,4为流速=1.0mL/min、T=45℃,5为流速=0.9mL/min,T=45℃。通过图3可以看出流动相流速和色谱柱柱温对样品的出峰时间和峰型有一定影响,其中流速为1.0mL/min,柱温45℃时峰型较好。2、不同载气流速、漂移管温度、不同仪器及不同色谱柱的影响取D-无水葡萄糖对照品适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖1000μg的溶液,即得对照品溶液。精密吸取上述对照品溶液5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件,分别在2.0L/min、2.5L/min和3.0mL/min的载气流速条件下,以及90℃、95℃和100℃的漂移管温度条件下,以及同一型号不同编号色谱柱条件下进行检测,记录色谱峰峰面积,结果如图4所示,图4是本发明实施例6提供的不同载气流速、漂移管温度和不同柱子的色谱图,图4中1为载气流速2.0L/min、飘移管温度95℃,2为载气流速2.5L/min、飘移管温度95℃,3为载气流速3.0L/min、飘移管温度95℃,4为载气流速2.0L/min、漂移管温度90℃,5为载气流速2.0L/min、漂移管温度100℃,6为载气流速2.0L/min、漂移管温度95℃,同一型号不同色谱柱子。通过图4可以看出载气流速、漂移管温度、不同仪器及不同色谱柱对样品的出峰时间和峰型影响不明显。实施例7精密度试验1、对照品溶液的制备:取D-无水葡萄糖对照品适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖0.5045mg、1.1099mg、1.7153mg的溶液,得低、中、高浓度对照品溶液。分别精密吸取上述低、中、高浓度对照品溶液各10μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,重复进样检测6次,计算进样精密度,结果如表8所示:表8低、中、高浓度对照品精密度试验结果通过表8可以看出,RSD分别为为1.16%、0.32%、0.30%。结果表明仪器精密度良好。实施例8稳定性试验1、对照品溶液的制备:取D-无水葡萄糖适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖0.8mg的溶液,即得对照品溶液。2、供试品溶液的制备:按照实施例2中所述待测样品液的制备方法制得供试品溶液。3、测试取同一供试品溶液(批号:140301),分别于第0h、2h、3h、5h、7h、9h、11h、13h、15h、17h、19h注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,每次进样5μL,记录色谱峰峰面积,并计算RSD值;另取同一对照品溶液,分别于第0h、2h、4h、6h、8h、10h、12h、19h、21h注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,每次进样5μL,记录色谱峰峰面积,并计算RSD值。结果见表9、10:表9供试品溶液稳定性试验结果表10对照品溶液稳定性试验结果通过表9和10可以看出,供试品溶液的RSD为2.21%,对照品溶液的RSD为1.61。说明供试品溶液在19小时内基本稳定,对照品溶液在21h内基本稳定。实施例9回收率试验取D-无水葡萄糖适量,精密称定,加80vol%乙醇制成每1mL含D-无水葡萄糖0.8mg的对照品溶液。取已知含量的样品(批号:140301,D-无水葡萄糖的含量为36.05wt%)约0.125g,精密称定,平行称取9份,3份一组,置具塞锥形瓶中,加入纯化水30mL。超声处理(40KHz,250W)30分钟,放冷,离心(11000r/min,15min),弃去上清液,沉淀加纯化水重新超声处理30min,离心(11000r/min,15min),将锥形瓶内剩余样品加入30mL纯化水少量多次的转移至离心管内,离心(11000r/min,15min),离心后沉淀加3mol/mL硫酸20mL少量多次转移至回流瓶内,各组分别加入D-无水葡萄糖对照品溶液,于沸水浴中回流3h,放冷,加35wt%氢氧化钠中和(PH为4.5-6.0),减压回收至一定体积,加乙醇溶解,使溶液乙醇浓度为70~80vol%,放冷,离心,溶液加80vol%乙醇定容至100mL,摇匀,即得加标待测样。分别精密吸取上述加标待测样各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到D-无水葡萄糖的含量,结果如表11所示。表11中,“称样量”是指制备供试品溶液时所称取的桂枝茯苓胶囊内容物的量,“含量”是指称取的内容物中理论上含有的D-无水葡萄糖糖的量,“加入量”是指加入的D-无水葡萄糖对照品的量,“测定值”是指供试品溶液中测得的D-无水葡萄糖的量,回收率=({含量+加入量)/测定值}×100%。表11D-无水葡萄糖加样回收率试验结果通过表11可以看出,评价回收率为100.11%,RSD为1.39%,说明本发明提供的方法准确性较高。实施例10桂枝茯苓胶囊中多糖含量的评价分别精密称取0.25g三批(批号:140701、140801、140901)桂枝茯苓胶囊的内容物,每批次样品称取两份,标记为①和②,按照实施例2中所述待测样品液的制备方法制得待测样品液。分别精密吸取上述待测样品液各5μL,注入高效液相色谱-ELSD检测器,按照实施例1中的色谱条件进行检测,记录色谱峰峰面积,根据所述峰面积与实施例1建立的D-无水葡萄糖的浓度-峰面积标准曲线,计算得到D-无水葡萄糖的含量,结果如表12所示:表12不同批次样品含量测定结果批号含量①含量②平均含量(%)RSD(%)14070139.3138.7639.030.9814080140.1438.8539.492.3214090138.6237.2737.952.52以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1