一种褐藻及其制品中海藻酸含量的检测方法与流程
本发明涉及分析化学技术领域,具体涉及一种褐藻及制品含用高效液相色谱法对海藻酸含量的检测方法。
背景技术:
海藻酸是从褐藻类植物中提取出来的碳水化合物,其为褐藻类植物的细胞壁填充物。组成上,海藻酸是以D-甘露糖醛酸和L-古罗糖醛酸两种糖醛酸聚合而成的非均匀长链高分子聚合物。一般而言,生物合成的海藻酸在结构上基本相似,但是海藻酸的D-甘露糖醛酸和L-古罗糖醛酸会依海藻种类、生态环境和季节有明显的不同,最终引起海藻酸的性质差异。
根据相关资料报道,海藻酸在制作冰激凌、冰牛奶、果酱、果冻和色拉等食品工业中可用作稳定剂和增稠剂;在纺织工业中可用作印花色浆,具有上色量高的特点,还可用作纺出人造纤维和止血纱布;在医疗卫生行业中可用作抗凝血剂和防治心血管药物,具有降低胆固醇含量、疏通血管、降低血液粘度、软化血管等作用,此外还可用作代血浆、止血剂等;在农业生产中可用于处理种子以及用于制作杀虫药分散剂、抗病毒喷洒剂等。因此,由于海藻酸广泛而显著的用途性,其生产及应用都引起了广泛关注。
目前,关于褐藻中海藻酸含量的测定方法主要有重量法、容量法、比色法和分光光度法,其中,比色法应用较广泛而分光光度法设备简单、费用低,但是上述方法均干扰多、精度不高。
为了克服现有褐藻中海藻酸含量的测定方法中的不足,实现方便、准确的检测目标分析物的目的,对生产中海藻酸含量进行有效监控,有必要研究一种应用高效液相色谱新的检测海藻及制品海藻酸含量的方法。
技术实现要素:
为了解决上述问题,本发明的目的是提供一种褐藻及其制品中海藻酸含量的检测方法。该检测方法是通过高效液相色谱法来实现对海藻酸含量的检测,具有高效快速、灵敏度高的优点,有效地解决了糖类物质没有紫外吸收的问题,可以准确地对海藻酸提取液进行定性和定量分析,且分离效果好,线性范围宽,具有针对性强、准确性、分离度高、分析快速、选择性好等优点,利于广泛应用。
本发明的目的是采用如下技术方案来实现的。
本发明提供一种褐藻及其制品中海藻酸含量的检测方法,该检测方法包括采用高效液相色谱法测定所述褐藻中海藻酸的含量,其中,所述高效液相色谱条件包括:
色谱柱:以十八烷基硅烷键合硅胶为填充剂的C18色谱柱;
流动相:乙腈-磷酸二氢钾缓冲液。
优选地,所述高效液相色谱法的条件包括:
所述C18色谱柱柱长为150mm,柱内径为4.6mm;
优选地,所述填充剂的粒径为5-10μm,优选为5μm;
所述流动相中所述磷酸二氢钾缓冲液的浓度为50mmol/L;优选地,所述乙腈-磷酸二氢钾缓冲液pH6.9;优选地,所述乙腈和磷酸二氢钾缓冲液体积比为10-25:75-90,优选为15:85。
优选地,所述高效液相色谱法的条件还包括:
色谱柱柱温:30-70℃,优选为40℃;
流动相流速:0.5-2.5mL/min,优选为0.5-1mL/min,进一步优选为1mL/min;
检测波长:250-300nm,优选为250nm。
优选地,所述高效液相色谱法的供试品溶液按照以下步骤来制备:
(1)取褐藻,经90-98%的浓硫酸于75-90℃下进行水解;
(2)在步骤(1)所得水解产物中加入氢氧化钠和1-苯基-3-甲基-5-吡唑啉酮,置于50-80℃下水浴中30-90min;
(3)在步骤(2)所得的溶液中加入三氯甲烷进行萃取,取上层清液,滤过;
优选地,所述步骤(1)中,所述水解时间为0.5-3h;
优选地,所述步骤(2)中加入氢氧化钠是指加入浓度为0.2-2mol/L的氢氧化钠溶液;
优选地,所述步骤(2)中加入1-苯基-3-甲基-5-吡唑啉酮是指加入浓度为0.5-2mol/L的1-苯基-3-甲基-5-吡唑啉酮溶液;
优选地,所述步骤(3)中所述萃取次数为2-4次;
优选地,所述每次萃取时间为20-60min;
优选地,所述步骤(3)中所述滤过是指使用0.20-0.60μm水系膜过滤,优选0.45μm水系膜;
优选地,所述步骤(3)在冰水浴中进行。
优选地,所述高效液相色谱法的供试品溶液按照以下步骤来制备:
(1)取褐藻1.25g,置于25ml容量瓶,加水溶解,定容,后移取1ml于20ml容量瓶中,加入0.5-5ml的90-98%的浓硫酸于75-90℃下进行水解0.5-3h,后冷却至室温,加入5ml水稀释后于82-90℃下水解30-90min;
(2)将步骤(1)所得水解产物置于冰水浴中冷却,经8mol/L的氢氧化钠溶液中和至pH值中性,后移置25ml容量瓶,定容,移取1ml于10ml容量瓶中,加入0.2-2mol/L氢氧化钠溶液0.5-5ml和0.5-2mol/L 1-苯基-3-甲基-5-吡唑啉酮溶液0.5-5ml,混合均匀,置于50-80℃下水浴中30-90min,后经0.3mol/L醋酸中和至pH值中性,再加水定容至10ml;
(3)冰水浴条件下,取步骤(2)所得溶液2ml置于20ml容量瓶,加入4ml三氯甲烷以萃取剩余的1-苯基-3-甲基-5-吡唑啉酮,萃取时间20-60min,弃去三氯甲烷层,反复萃取2-4次,取上层清液,经0.45μm水系膜过滤。
优选地,所述高效液相色谱法的对照品溶液按照以下步骤来制备:
(1)取海藻酸钠,经90-98%的浓硫酸于75-90℃下进行水解;
(2)在步骤(1)所得水解产物中加入氢氧化钠和1-苯基-3-甲基-5-吡唑啉酮,置于50-80℃下水浴中30-90min;
(3)在步骤(2)所得溶液中加入三氯甲烷进行萃取,取上层清液,滤过;
优选地,所述步骤(1)中所述水解时间为0.5-3h;
优选地,所述步骤(2)中加入氢氧化钠是指加入浓度为0.2-2mol/L的氢氧化钠溶液;
优选地,所述步骤(2)中加入1-苯基-3-甲基-5-吡唑啉酮是指加入浓度为0.5-2mol/L的1-苯基-3-甲基-5-吡唑啉酮溶液;
优选地,所述步骤(3)中所述萃取次数为2-4次;优选地,所述每次萃取时间为20-60min;
优选地,所述步骤(3)中所述滤过是指使用0.20-0.60μm水系膜过滤,优选0.45μm水系膜;
优选地,所述步骤(3)在冰水浴中进行。
优选地,所述高效液相色谱法的对照品溶液按照以下步骤来制备:
(1)分别取海藻酸钠5mg、10mg、15mg、20mg、25mg,再分别加1ml水溶解,定容,后分别移取1ml于20ml容量瓶中,加入0.5-5ml的90-98%的浓硫酸于75-90℃下进行水解0.5-3h,后冷却至室温,加入5ml水稀释后于82-90℃下水解30-90min;
(2)分别将步骤(1)所得水解产物置于冰水浴中冷却,经8mol/L的氢氧化钠溶液中和至pH值中性,后分别移置25ml容量瓶,定容,分别移取1ml于10ml容量瓶中,加入0.2-2mol/L氢氧化钠溶液0.5-5ml和0.5-2mol/L1-苯基-3-甲基-5-吡唑啉酮溶液0.5-5ml,混合均匀,置于50-80℃下水浴中30-90min,后经0.3mol/L醋酸中和至pH值中性,再加水定容至10ml;
(3)冰水浴条件下,分别取步骤(2)所得溶液2ml置于20ml容量瓶,分别加入4ml三氯甲烷以分别萃取剩余的1-苯基-3-甲基-5-吡唑啉酮,萃取时间20-60min,弃去三氯甲烷层,反复2-4次,取上层清液,经0.45μm水系膜过滤,分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
优选地,所述褐藻中海藻酸含量的检测方法包括以下步骤:
(a)制备供试品溶液:
(a-1)取褐藻1.25g,置于25ml容量瓶,加水溶解,定容,后移取1ml于20ml容量瓶中,加入0.5-5ml的90-98%的浓硫酸于75-90℃下进行水解0.5-3h,后冷却至室温,加入5ml水稀释后于82-90℃下水解30-90min;
(a-2)将步骤(a-1)获得的水解产物置于冰水浴中冷却,经8mol/L的氢氧化钠溶液中和至pH值中性,后移置25ml容量瓶,定容,移取1ml于10ml容量瓶中,加入0.2-2mol/L氢氧化钠溶液0.5-5ml和0.5-2mol/L 1-苯基-3-甲基-5-吡唑啉酮溶液0.5-5ml,混合均匀,置于50-80℃下水浴中30-90min,后经0.3mol/L醋酸中和至pH值中性,再加水定容至10ml;
(a-3)冰水浴条件下,取步骤(a-2)获得的溶液2ml置于20ml容量瓶,加入4ml三氯甲烷以萃取剩余的1-苯基-3-甲基-5-吡唑啉酮,萃取时间20-60min,弃去三氯甲烷层,反复萃取2-4次,取上层清液,经0.45μm水系膜过滤。
(b)制备对照品溶液:
(b-1)分别取海藻酸钠5mg、10mg、15mg、20mg、25mg,再分别加1ml水溶解,定容,后分别移取1ml于20ml容量瓶中,加入0.5-5ml的90-98%的浓硫酸于75-90℃下进行水解0.5-3h,后冷却至室温,加入5ml水稀释后于82-90℃下水解30-90min;
(b-2)分别将步骤(b-1)所得的水解产物置于冰水浴中冷却,经8mol/L的氢氧化钠溶液中和至pH值中性,后分别移置25ml容量瓶,定容,分别移取1ml于10ml容量瓶中,加入0.2-2mol/L氢氧化钠溶液0.5-5ml和0.5-2mol/L 1-苯基-3-甲基-5-吡唑啉酮溶液0.5-5ml,混合均匀,置于50-80℃下水浴中30-90min,后经0.3mol/L醋酸中和至pH值中性,再加水定容至10ml;
(b-3)冰水浴条件下,分别取步骤(b-2)所得的溶液2ml置于20ml容量瓶,分别加入4ml三氯甲烷以分别萃取剩余的1-苯基-3-甲基-5-吡唑啉酮,萃取时间20-60min,弃去三氯甲烷层,反复2-4次,取上层清液,经0.45μm水系膜过滤,分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
(c)采用高效液相色谱法测定所述海藻酸的含量:
取对照品溶液和供试品溶液注入高效液相色谱仪进行测定,所述高效液相色谱包括:
色谱柱:以十八烷基硅烷键合硅胶为填充剂的C18色谱柱,优选地,所述色谱柱柱长为150mm,柱内径为4.6mm;优选地,所述色谱柱中填充剂的粒径为5-10μm,优选5μm;
色谱柱柱温:30-70℃,优选为40℃;
流动相流速:0.5-2.5mL/min,优选为0.5-1mL/min,进一步优选为1mL/min;
检测波长:250-300nm,优选为250nm;
所述流动相中所述磷酸二氢钾缓冲液的浓度为50mmol/L;优选地,所述乙腈-磷酸二氢钾缓冲液pH6.9;优选地,所述乙腈和磷酸二氢钾缓冲液体积比为10-25:75-90,优选15:85。
与现有技术相比,本发明提供的检测海藻酸含量的方法具有以下有益效果:
(1)本发明将海藻酸含量测定的指标选定为D-甘露糖醛酸和L-古罗糖醛酸,此二者为海藻酸中有代表性的主要成分,含量高、性质稳定,作为标示性成分用于控制海藻酸含量具有较好的专属性和质量相关性;
(2)相对于紫外分光光度法、薄层扫描法等而言,本发明的高效液相色谱法具有分辨率高、灵敏性高、重现性好、选择性好、结果误差小等优点;
(3)本发明的检测方法对供试品和对照品的处理快速、方便、易操作;
(4)本发明的检测方法实现了对海藻酸进行定性和定量分析,且分离效果好,线性范围宽,具有针对性强、准确性、分离度高、分析快速、选择性好等优点,利于广泛应用,填补了海藻酸含量分析方面的研究空白;
(5)不同的色谱条件、不同的供试品和对照品的处理方法对于海藻酸含量的检测结果会有很大影响,本发明经筛选优选获得的高效液相色谱法检测条件和步骤可以准确、简便地测定海藻酸的含量。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
图1为海藻酸钠对照品的色谱图,其中1号峰为L-古罗糖醛酸,2号峰为D-甘露糖醛酸;
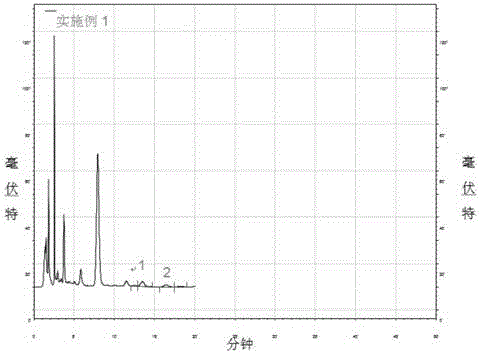
图2为实施例1液相检测样品中海藻酸的色谱图;
图3为实施例4液相检测样品中海藻酸的色谱图;
图4为实施例5液相检测样品中海藻酸的色谱图。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。
以下各实施例中所使用的供试品来源于北京雷力海洋生物新产业股份有限公司自制,对照品海藻酸钠采购于SIGMA公司。
高效液相色谱仪及检测器采购于美国SSI公司;C18色谱柱采购于岛津公司。
实施例1
1、制备供试品溶液:
(1)褐藻水解:
准确称取褐藻1.25g(精确到0.0001g),置于25ml容量瓶中,加水充分溶解,定容,然后精密移取1ml置于20ml水解瓶中(液体直接精密分取1ml),加1.0ml浓度为98%的浓硫酸,然后于82℃水浴中水解30min取出后冷却至室温,加5ml水稀释后再置于82℃水浴中继续水解30min;
(2)供试品的衍生:
取出步骤(1)水解产物放置冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入0.3mol/L的氢氧化钠溶液1ml和0.5mol/L的PMP(1-苯基-3-甲基-5-吡唑啉酮)溶液1ml,混匀后于70℃水浴中衍生70min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)供试品的预处理:
在冰水浴中,取步骤(2)所得溶液2ml置于20ml水解瓶中,加入4ml三氯甲烷涡旋混匀,放置30min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取两次,取上层清液经0.45μm水系膜过滤。
2、制备对照品溶液:
(1)海藻酸钠水解:
分别准确称取海藻酸钠5mg、10mg、15mg、20mg、25mg于水解瓶中,再分别加1ml蒸馏水温热充分溶解,定容,后分别移取1ml于20ml容量瓶中,加入1ml浓度为98%的浓硫酸,然后于82℃水浴中水解30min取出后冷却至室温,加5ml水稀释后再置于82℃水浴中继续水解30min;
(2)对照品的衍生:
分别将步骤(1)获得的各水解产物置于冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后分别转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入0.3mol/L的氢氧化钠溶液1ml和0.5mol/L的PMP溶液1ml,混匀后于70℃水浴中衍生70min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)对照品的预处理:
在冰水浴中,分别取步骤(2)所得的各溶液2ml置于20ml水解瓶中,分别加入4ml三氯甲烷涡旋混匀,放置30min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取2次,取上层清液经0.45μm水系膜过滤;分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
3、高效液相色谱仪测定:
测定对照品溶液和供试品溶液的紫外吸收响应值,根据峰保留时间定性,并根据峰面积外标法进行定量;
色谱条件:C18色谱柱,150mm×4.6mm,5μm;
柱温:40℃;
流速:1.0mL/min;
紫外检测器波长:250nm;
进样体积:20μL;
流动相:乙腈与50mmol/L的磷酸二氢钾缓冲液的体积比为15:85。
4、标准曲线的建立:
采用外标法对海藻酸钠不同浓度(即20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL)的对照品溶液从低浓度到高浓度依次进液相色谱分析,得到相应谱图,采用峰面积定量,以峰面积(积分值)对质量浓度(mg/L)求得线性回归方程,验证标准曲线方程和相关系数,确定线性范围为0.02-0.16mg/mL,线性回归方程和相关系数分别为:y=35135x-29814和r=0.9994。
5、本发明海藻酸含量的检测方法的方法学考察
通过检测褐藻中海藻酸供试品溶液进行方法相对标准偏差和加标回收率考察,对于海藻酸供试品溶液的加标水平分别为25mg/g、100mg/g、300mg/g,进行3次加标测定分析,结果如表1和表2所示,三次平行测定值的相对标准偏差在2.38-3.39之间,回收率在80%至110%之间,变异系数在2%至15%之间,结果重复性好,添加回收率高,方法的准确度和精密度可以满足海藻酸供试品溶液含量测定的要求。采用空白样品标准添加的方法确定方法的检出限为10mg/g,定量限为20mg/g。
表1:方法相对标准偏差考察
表2:加标回收率考察
6、计算方法:
褐藻中海藻酸含量为古罗糖醛酸含量和甘露糖醛酸含量之和。
式中:
X1:褐藻中古罗糖醛酸百分含量;
X2:褐藻中甘露糖醛酸百分含量;
C1:Sigma海藻酸钠中古罗糖醛酸百分含量;
C2:Sigma海藻酸钠中甘露糖醛酸百分含量;
S1:海藻酸钠对照品溶液中古罗糖醛酸峰面积;
S2:海藻酸钠对照品溶液中甘露糖醛酸峰面积;
S3:褐藻中古罗糖醛酸峰面积;
S4:褐藻中甘露糖醛酸峰面积;
M1:海藻酸钠质量单位为克(g);
M3:褐藻质量单位为克(g);
V1:对照品海藻酸钠定容体积,单位为毫升(mL);
V2:供试品褐藻溶液定容体积,单位为毫升(mL)。
实施例2
1、制备供试品溶液:
(1)褐藻水解:
准确称取褐藻1.25g(精确到0.0001g),置于25ml容量瓶中,加水充分溶解,定容,然后精密移取1ml置于20ml水解瓶中(液体直接精密分取1ml),加0.5ml浓度为90%的浓硫酸,然后于75℃水浴中水解60min取出后冷却至室温,加5ml水稀释后再置于75℃水浴中继续水解60min;
(2)供试品的衍生:
取出步骤(1)水解产物放置冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入0.2mol/L的氢氧化钠溶液0.5ml和1mol/L的PMP溶液0.5ml,混匀后于50℃水浴中衍生30min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)供试品的预处理:
在冰水浴中,取步骤(2)所得溶液2ml置于20ml水解瓶中,加入4ml三氯甲烷涡旋混匀,放置20min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取2次,取上层清液经0.2μm水系膜过滤。
2、制备对照品溶液:
(1)海藻酸钠水解:
分别准确称取海藻酸钠5mg、10mg、15mg、20mg、25mg于水解瓶中,再分别加1ml蒸馏水温热充分溶解,定容,后分别移取1ml于20ml容量瓶中,加入0.5ml浓度为90%的浓硫酸,然后于75℃水浴中水解60min取出后冷却至室温,加5ml水稀释后再置于75℃水浴中继续水解60min;
(2)对照品的衍生:
分别将步骤(1)获得的各水解产物置于冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后分别转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入0.2mol/L的氢氧化钠溶液0.5ml和1mol/L的PMP溶液0.5ml,混匀后于50℃水浴中衍生30min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)对照品的预处理:
在冰水浴中,分别取步骤(2)所得的各溶液2ml置于20ml水解瓶中,分别加入4ml三氯甲烷涡旋混匀,放置20min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取2次,取上层清液经0.20μm水系膜过滤;分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
3、高效液相色谱仪测定:
测定对照品溶液和供试品溶液的紫外吸收响应值,根据峰保留时间定性,并根据峰面积外标法进行定量;
色谱条件:C18色谱柱,150mm×4.6mm,5μm;
柱温:30℃;
流速:0.5mL/min;
紫外检测器波长:280nm;
进样体积:20μL;
流动相:乙腈与50mmol/L的磷酸二氢钾缓冲液的体积比为10:90。
实施例3
1、制备供试品溶液:
(1)褐藻水解:
准确称取褐藻1.25g(精确到0.0001g),置于25ml容量瓶中,加水充分溶解,定容,然后精密移取1ml置于20ml水解瓶中(液体直接精密分取1ml),加1.0ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)供试品的衍生:
取出步骤(1)水解产物放置冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入2mol/L的氢氧化钠溶液5ml和2mol/L的PMP溶液5ml,混匀后于80℃水浴中衍生90min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)供试品的预处理:
在冰水浴中,取步骤(2)所得溶液2ml置于20ml水解瓶中,加入4ml三氯甲烷涡旋混匀,放置60min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取4次,取上层清液经0.60μm水系膜过滤;
2、制备对照品溶液:
(1)海藻酸钠水解:
分别准确称取海藻酸钠5mg、10mg、15mg、20mg、25mg于水解瓶中,再分别加1ml蒸馏水温热充分溶解,定容,后分别移取1ml于20ml容量瓶中,加入5ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)对照品的衍生:
分别将步骤(1)获得的各水解产物置于冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后分别转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入2mol/L的氢氧化钠溶液5ml和2mol/L的PMP溶液5ml,混匀后于80℃水浴中衍生90min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)对照品的预处理:
在冰水浴中,分别取步骤(2)所得的各溶液2ml置于20ml水解瓶中,分别加入4ml三氯甲烷涡旋混匀,放置60min萃取多余的PMP,然后弃去三氯甲烷层,反复萃取4次,取上层清液经0.60μm水系膜过滤;分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
3、高效液相色谱仪测定:
测定对照品溶液和供试品溶液的紫外吸收响应值,根据峰保留时间定性,并根据峰面积外标法进行定量;
色谱条件:C18色谱柱,150mm×4.6mm,5μm;
柱温:70℃;
流速:0.5mL/min;
紫外检测器波长:300nm;
进样体积:20μL;
流动相:乙腈与50mmol/L的磷酸二氢钾缓冲液的体积比为25:75。
实施例4
1、制备供试品溶液:
(1)褐藻水解:
准确称取褐藻1.25g(精确到0.0001g),置于25ml容量瓶中,加水充分溶解,定容,然后精密移取1ml置于20ml水解瓶中(液体直接精密分取1ml),加1.0ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)供试品不进行衍生。
2、制备对照品溶液:
(1)海藻酸钠水解:
分别准确称取海藻酸钠5mg、10mg、15mg、20mg、25mg于水解瓶中,再分别加1ml蒸馏水温热充分溶解,定容,后分别移取1ml于20ml容量瓶中,加入5ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)对照品不进行衍生:
3、高效液相色谱仪测定:
测定对照品溶液和供试品溶液的紫外吸收响应值,根据峰保留时间定性,并根据峰面积外标法进行定量;
色谱条件:C18色谱柱,150mm×4.6mm,5μm;
柱温:70℃;
流速:0.5mL/min;
紫外检测器波长:300nm;
进样体积:20μL;
流动相:乙腈与50mmol/L的磷酸二氢钾缓冲液的体积比为25:75;
4、数据结果:
见图3,供试品和对照品都未出现相应峰,因此供试品和对照品不衍生,紫外检测器几乎无吸收,因此不衍生的结论是不可行的。
实施例5
1、制备供试品溶液:
(1)褐藻水解:
准确称取褐藻1.25g(精确到0.0001g),置于25ml容量瓶中,加水充分溶解,定容,然后精密移取1ml置于20ml水解瓶中(液体直接精密分取1ml),加1.0ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)供试品的衍生:
取出步骤(1)水解产物放置冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入2mol/L的氢氧化钠溶液5ml和2mol/L的对氨基苯甲酸(p-AMBA)溶液5ml,混匀后于80℃水浴中衍生90min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)供试品的预处理:
在冰水浴中,取步骤(2)所得溶液2ml置于20ml水解瓶中,加入4ml三氯甲烷涡旋混匀,放置60min萃取多余的p-AMBA,然后弃去三氯甲烷层,反复萃取4次,取上层清液经0.60μm水系膜过滤。
2、制备对照品溶液:
(1)海藻酸钠水解:
分别准确称取海藻酸钠5mg、10mg、15mg、20mg、25mg于水解瓶中,再分别加1ml蒸馏水温热充分溶解,定容,后分别移取1ml于20ml容量瓶中,加入5ml浓度为95%的浓硫酸,然后于90℃水浴中水解90min取出后冷却至室温,加5ml水稀释后再置于90℃水浴中继续水解90min;
(2)对照品的衍生:
分别将步骤(1)获得的各水解产物置于冰水浴中冷却,然后用8mol/L的氢氧化钠溶液中和,调pH值至中性,然后分别转移至25ml容量瓶定容,取1ml溶液于10ml容量瓶中加入2mol/L的氢氧化钠溶液5ml和2mol/L的对氨基苯甲酸(p-AMBA)溶液5ml,混匀后于80℃水浴中衍生90min,后放至室温,用0.3mol/L的醋酸中和,然后用水定容至10ml,备用;
(3)对照品的预处理:
在冰水浴中,分别取步骤(2)所得的各溶液2ml置于20ml水解瓶中,分别加入4ml三氯甲烷涡旋混匀,放置60min萃取多余的p-AMBA,然后弃去三氯甲烷层,反复萃取4次,取上层清液经0.60μm水系膜过滤;分别得到浓度为20μg/mL、40μg/mL、60μg/mL、80μg/mL、100μg/mL的对照品溶液。
3、高效液相色谱仪测定:
测定对照品溶液和供试品溶液的紫外吸收响应值,根据峰保留时间定性,并根据峰面积外标法进行定量;
色谱条件:C18色谱柱,150mm×4.6mm,5μm;
柱温:70℃;
流速:0.5mL/min;
紫外检测器波长:300nm;
进样体积:20μL;
流动相:乙腈与50mmol/L的磷酸二氢钾缓冲液的体积比为25:75。
4、数据结果:
由图4可以看出,供试品和对照品出现的峰高较低,峰不对称,因此供试品和对照品用p-AMBA衍生分离效果不好。
- 还没有人留言评论。精彩留言会获得点赞!