基于低共熔溶剂-无机盐的双水相体系分析水样中农药残留的方法与流程
本发明涉及水样中农药残留的分析方法,具体涉及一种基于低共熔溶剂-无机盐的双水相体系分析水样中三唑类农药残留的方法。
(二)
背景技术:
环境中农药的浓度往往非常低(ppt-ppb级),并且不同基质(蔬菜和土壤样品)干扰物质的差异很大,样品前处理是农药残留分析过程中最重要、最关键的步骤,不仅要对样品进行高倍富集,还需有效除去干扰物质。气相色谱-质谱联用和液相色谱-质谱联用兼有高的检测灵敏度和强大的定性能力,已经成为农产品和环境样品中三唑类农药残留检测的主流分析仪器。经典的样品前处理技术存在操作繁琐耗时,需要使用大量的有毒或有害的有机溶剂等缺点,所以,近年来人们致力于建立无污染或少污染的绿色分析化学技术。
一个完整的样品分析过程,从采样开始到写出分析报告,大致可分为4个步骤:①样品采集;②样品前处理;③分析测定;④数据处理与报告结果。这4个步骤中各步所需的时间相差很大,它们占全部分析时间的比例分别为:样品采集6.0%,样品前处理61.0%,分析测定6.0%,数据处理与报告27.0%,其中样品前处理所需的时间最长,约占整个分析时间的三分之二。传统的前处理技术如索式萃取、液-液萃取、水蒸气蒸馏萃取等,这些技术存在诸如萃取效率低或处理时间长等缺点,且很多方法中大量使用的有毒有害有机溶剂会造成环境污染。
beijeronck在1896年将琼脂水溶液与可溶性淀粉或明胶水溶液混合,发现了双水相现象。首次实现对双水相体系进行应用是在20世纪60年代,瑞典伦德大学的albertsson将双水相体系成功用于分离叶绿素,解决了蛋白质变性和沉淀的问题。1979年德国kula等人将双水相萃取分离技术应用于生物酶的分离,为双水相在生物蛋白质、酶分离纯化等的应用奠定了基础。迄今为止,双水相成功地应用在生物医药工程,天然产物分离纯化,金属离子分离等方面。
将2种不同的水溶性聚合物水溶液(或聚合物与一定浓度的盐溶液)混合时,当聚合物浓度(或盐的浓度)达到一定值,体系会自然分成互不相溶的两相,这就是双水相体系。双水相体系的形成主要是由于高聚物之间的不相溶性,一般认为只要两聚合物水溶液的憎水程度有所差异混合时就可发生相分离,且憎水程度相差越大,相分离倾向也就越大。
双水相萃取的原理与传统的水-有机相萃取的原理相似,都是依据物质在两相间的选择性分配,但萃取体系的性质不同。当物质进入双水相体系后,由于表面性质、电荷作用和各种力(如憎水键、氢键和离子键等)的存在和环境的影响,使其在上、下相中的浓度不同。对于某一物质,只要选择合适的双水相体系,控制一定的条件,就可以得到合适的分配系数,从而达到分离纯化之目的。
但是与传统萃取相比,双水相有其独特之处:(1)两相的溶剂都是水,上相和下相的含水量高达70%~90%(w/w),不存在有机溶剂残留问题。条件很温和,常温常压操作,不会引起生物活性物质失活或变性;(2)两相界面张力小,仅为10-6~10-4n/m(普通体系为10-3~10-2n/m),双水相的两相差别(如密度、折射率)相差很小,萃取时两相能够高度分散,传质速度快,但也引起乳化现象;(3)溶剂对目标组分选择性强,大量杂质能与所有固体物质一同除去,使分离过程简化,易于工业放大和连续操作;(4)分相时间短,常温常压下自然分相时间一般为5~10min;(5)目标产物的分配系数一般大于3,大部分情况下目标产物的收率较高;(6)聚合物的浓度、无机盐的种类和浓度,以及体系的ph值等多种因素都可以对被萃取物质在两相的分配产生影响,因此可以利用多种手段来使萃取达到最佳条件;(7)该体系可以处理以固体微粒形式出现的样品。
传统萃取分离技术的萃取剂是挥发性强、具有毒性的有机溶剂,其对环境造成的危害越来越受到人们的关注,离子液体双水相体系萃取分离是近年来出现的一种新型体系分离,离子液体双水相体系的研究始于2002年,dupont等在合成离子液体1-丁基-3-甲基咪唑四氟硼酸盐时意外发现kcl和[c4mim][bf4]之间存在盐析效应。rogers等对这种盐析效应做了进一步研究,并提出了“离子液体双水相”这一新概念。离子液体双水相通常是指由一种亲水性离子液体、一种无机盐(磷酸盐,碳酸盐,氢氧化物等)和水形成的体系,这个体系一般会形成上相富含离子液体,而下相富含盐的两相,所以称为离子液体双水相,其在萃取分离方面有良好的应用前景。
相比于传统的分子溶剂,离子液体具有蒸汽压极低、不可燃、溶解性能强和黏度可变等特点,已在许多领域得到广泛应用。然而,离子液体的合成成本较高且可生物降解性较差,对环境造成一定的影响,这些缺点限制了离子液体的大规模生产和进一步广泛应用。abbott课题组在2003年首次发现了一种由氯化胆碱和尿素组成的低共熔溶剂(deepeutecticsolvents,des),其合成过程原子利用率达100%,具有蒸汽压低、无毒性、可生物降解等独特的物理化学性质,并且可以通过选择合适的组成配比来调节其性能,是一种新型的绿色溶剂。des制备方法简单,通常只需将一定比例的氢键供体(hbd,如羧酸类、醇类等)和氢键受体(hba,如季铵盐、季鏻盐等)在一定温度下搅拌即可完成制备。
低共熔溶剂具有离子液体的优良性质,但比离子液体廉价、容易制备且环境友好,des的这些特性大大拓宽了它的应用领域。低共熔溶剂具有良好的溶解性质,对许多金属氧化物有较好的溶解性能,包括cuo、zno、mno2、fe3o4等,对一些气体如co2、so2等具有良好的溶解性。
(三)
技术实现要素:
针对现有技术中存在的不足,本发明旨在建立一种新型的低共熔溶剂-无机盐的双水相体系,并结合涡旋辅助萃取以及离心分离的技术用于环境水样中农药残留测定。该方法利用离心管作为萃取装置,通过涡旋的方式使得目标分析物在低共熔溶剂和无机盐溶液中进行分配转移,然后通过离心的方式,通过形成双水相的方式实现相分离。
为了实现上述目的,本发明提供了一种基于低共熔溶剂-无机盐的双水相体系萃取的前处理方法并且用于分析水样中的农药残留,本发明方法所需设备简单,操作简便且操作时间短,可以适用于不同体积样品的前处理操作,同时具有批量样品同时进行操作的优点。
本发明的技术方案如下:
一种基于低共熔溶剂-无机盐的双水相体系分析水样中农药残留的方法,所述水样中的农药为三唑类农药,所述三唑类农药为下列化合物中的至少一种:腈菌唑、戊唑醇、苯醚甲环唑;
所述方法包括如下步骤:
(1)样品前处理
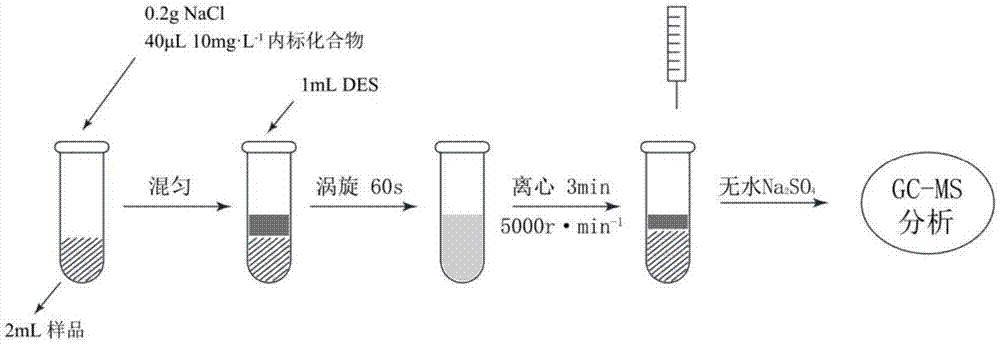
样品前处理:首先,取待测水样于离心管中,加入固体无机盐和内标化合物氟环唑,待固体溶解完全后,再向离心管中加入低共熔溶剂,涡旋(2500rpm,60s)使其乳化,离心(5000rpm,3min),取上层液体,干燥后即完成前处理过程;
所述固体无机盐的质量用量以待测水样的体积计为0.1g/ml;所述固体无机盐为na2so4、na2co3、nacl、k2co3或kcl,优选nacl;
所述内标化合物氟环唑的质量用量以待测水样的体积计为200μg/l;
所述待测水样与低共熔溶剂的体积比为1:0.2~1;
所述低共熔溶剂选自下列之一:
氯化胆碱(氢键受体):对氯苯酚(氢键供体)物质的量之比1:2的低共熔溶剂;
氯化胆碱(氢键受体):苯甲醇(氢键供体)物质的量之比1:3的低共熔溶剂;
氯化胆碱(氢键受体):苯甲醇(氢键供体)物质的量之比1:4的低共熔溶剂;
氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:2的低共熔溶剂;
氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:3的低共熔溶剂;
氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:4的低共熔溶剂;
所述低共熔溶剂的制备方法为:将氢键受体、氢键供体混合,升温至80℃搅拌2h,之后降至室温即得;
优选低共熔溶剂为氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:3的低共熔溶剂;
(2)样品检测
将经过前处理的样品注入气相色谱-质谱联用仪进行分析,得到样品气相色谱质谱总离子流图;
气相色谱条件为:色谱柱db-5ms(长度30m×内径0.25mm×膜厚0.25μm),初始柱温为180℃,在此温度下保持1min,然后以5℃/min升温至200℃,保持1min,之后以2℃/min升温至220℃,最后以10℃/min升温至290℃,保持6min;高纯氦气(99.999%)为载气,流速为1.0ml/min;进样口温度:280℃;进样量:1.0μl,不分流模式进样;
质谱条件为:选择离子扫描模式,离子阱温度为180℃,传输线温度为250℃,岐管温度为50℃,电子碰撞能量为70ev;
(3)建立标准曲线
取腈菌唑、戊唑醇、苯醚甲环唑的标准物质,以甲醇为溶剂配制混合标准储备液(-4℃避光保存),所得混合标准储备液经稀释(用去离子水)得到标准曲线工作溶液,将所得标准曲线工作溶液按照步骤(1)所述前处理方法进行前处理之后(即将步骤(1)中待测水样替换为标准曲线工作溶液),再于步骤(2)中的检测条件下注入气相色谱-质谱联用仪进行分析,得到标准物质的气相色谱质谱总离子流图,以气相色谱质谱总离子流图中的标准物质特征峰面积与内标化合物特征峰面积的比值为纵坐标,标准曲线工作溶液中的标准物质浓度为横坐标,绘制标准曲线;
各标准物质在标准曲线工作溶液中的浓度范围如下:
腈菌唑20~2000μg/l;戊唑醇20~2000μg/l;苯醚甲环唑20~1000μg/l;(内标化合物氟环唑200μg/l)
各标准物质在气相色谱质谱总离子流图中特征峰如下:
腈菌唑13.15min;戊唑醇17.86min;苯醚甲环唑25.20min;(内标化合物氟环唑18.42min)
(4)获取样品中农药残留的定性和定量结果
通过样品气相色谱质谱总离子流图和标准物质气相色谱质谱总离子流图的对照,对样品中所含三唑类农药进行定性;
将步骤(2)所得样品气相色谱质谱总离子流图中的三唑类农药特征峰面积值与内标化合物特征峰面积的比值代入步骤(3)所得标准曲线中,计算获得样品中三唑类农药的含量。
与现有技术相比,本发明的有益效果为:
1、本发明提供了提取环境水样中农药残留的有效方法;
2、首次将新型低共熔溶剂引入双水相体系中,克服了大多数的低共熔溶剂都具有一定的水溶性而使得其在样品基质上的应用受到了限制的问题;
3、采用低共熔溶剂作为萃取剂,减少对环境的污染;
4、萃取装置易于清洗从而减少样品和溶剂的残留;
5、应用本发明方法能结合实际,可以适用于不同体积样品的前处理操作,同时具有批量样品同时进行操作的优点,为环境污染物和农药残留等环境和食品安全问题提供了一种检测手段。
(四)附图说明
图1为本发明建立的基于低共熔溶剂-无机盐双水相体系的萃取过程示意图;
图2a为按照实施例1方法对双水相形成组分中低共熔溶剂的种类进行优化的结果;
des-1:氯化胆碱(氢键受体):对氯苯酚(氢键供体)物质的量之比1:2的低共熔溶剂;
des-2:氯化胆碱(氢键受体):苯甲醇(氢键供体)物质的量之比1:3的低共熔溶剂;
des-3:氯化胆碱(氢键受体):苯甲醇(氢键供体)物质的量之比1:4的低共熔溶剂;
des-4:氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:2的低共熔溶剂;
des-5:氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:3的低共熔溶剂;
des-6:氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:4的低共熔溶剂;
图2b为按照实施例1方法对双水相形成组分中无机盐的种类进行优化的结果;
图2c为按照实施例1方法对双水相形成组分中无机盐添加量进行优化的结果;
图3a为按照实施例1方法得到的空白水样以及不同浓度加标的空白水样的气相色谱质谱总离子流图;
其中的(a)、(b)、(c)、(d)分别为空白水样,加标浓度为20μg/l的空白水样,加标浓度为200μg/l的空白水样以及加标浓度为1000μg/l的空白水样,其中标注的1、2、3、4号峰分别为腈菌唑、戊唑醇、氟环唑、苯醚甲环唑;
图3b为实施例1中实际样品的气相色谱质谱总离子流图;
其中标注的1、2、3、4号峰分别为腈菌唑、戊唑醇、氟环唑、苯醚甲环唑。
(五)具体实施方式
下面通过具体实施例对本发明作进一步描述,但本发明的保护范围并不仅限于此。
实施例1:环境水样中三唑类农药残留的检测(内标法)
(1)样品前处理及检测
低共熔溶剂为氯化胆碱(氢键受体):苯酚(氢键供体)物质的量之比1:3的混合物。制备:称取35g干燥过的氯化胆碱,70.5g苯酚,置于500ml圆底烧瓶中,并于80℃条件下持续搅拌2h直至形成澄清均一的液体溶剂。
首先,量取待测水样2ml于7ml离心管中,加入固体无机盐nacl0.2g和10mg·l-1的内标化合物氟环唑40μl,待混合均匀后,再向离心管中加入1ml上述合成的低共熔溶剂,在ikavortex天才3圆周振荡器中以2500rpm涡旋60s,可以明显观察到乳浊液的形成。然后在5000r·min-1下离心3min,用气相色谱进样针取出上层有机层,转移至装有无水硫酸钠的pcr管中进行除水。吸取干燥后的有机层1μl进入气相色谱-质谱联用仪进行分析。
气相色谱条件为:色谱柱db-5ms(长度30m×内径0.25mm×膜厚0.25μm),初始柱温为180℃,在此温度下保持1min,然后以5℃min-1升温至200℃,保持1min,之后以2℃min-1升温至220℃,最后以10℃min-1升至290℃,保持6min;高纯氦气(99.999%)为载气,流速为1.0mlmin-1;进样口温度:280℃;进样量:1.0μl,不分流模式进样。
质谱条件为:选择离子扫描模式,离子阱温度为180℃,传输线温度为250℃,岐管温度为50℃,电子碰撞能量为70ev;
(2)建立标准曲线
分别准确称取标准物质腈菌唑0.1038g、戊唑醇0.1031g、苯醚甲环唑0.1032g置于烧杯中,先溶于100ml甲醇得到混标储备液,取所得混标储备液1ml,用去离子水稀释至100ml容量瓶中;再取稀释后的溶液1ml,用去离子水分别稀释500、200、100、50、20、10、5倍,得到7个标准曲线工作溶液。然后按照步骤(1)进行前处理并注入气相色谱-质谱联用仪进行分析,得到标准物质气相色谱质谱总离子流图,以气相色谱质谱总离子流图中的标准物质特征峰面积与内标物质特征峰面积的比值为纵坐标,标准曲线工作溶液中的标准物质浓度为横坐标,绘制标准曲线。
各标准物质在气相色谱图中特征峰如下:
腈菌唑13.15min;戊唑醇17.86min;苯醚甲环唑25.20min;内标化合物氟环唑18.42min。
分别得到如下标准曲线:
腈菌唑:y=13.64x-0.2327
戊唑醇:y=3.054x+0.2205
苯醚甲环唑:y=26.40x+0.5592
(3)获取水样中农药残留的定性和定量结果
通过步骤(1)实际样品气相色谱质谱总离子流图和标准物质气相色谱质谱总离子流图的对照,对样品中所含三唑类农药进行定性;
将步骤(1)实际样品气相色谱质谱总离子流图中各三唑类农药的特征峰面积值与内标化合物特征峰面积的比值代入步骤(2)所得标准曲线中,计算获得水样中各三唑类农药的含量,结果如下:腈菌唑6.27μg·l-1、戊唑醇3.07μg·l-1、苯醚甲环唑4.53μg·l-1。
(4)方法评估
在步骤(1)~(3)的条件下,对线性范围、相关系数、相对标准偏差、检测限、定量限等参数进行了考察。标准曲线在7个不同浓度点进行三次平行测定后得到,结果列在表1中。如表1所示,三种三唑类农药的线性范围为腈菌唑20-2000μg·l-1、戊唑醇20-2000μg·l-1和苯醚甲环唑20-1000μg·l-1,相应的相关系数为0.996-0.999。当信噪比(s/n)分别为3和10得到的检测限和定量限分别为0.78-1.10μg·l-1和2.34-3.30μg·l-1。通过三种浓度(20、200、1000μg·l-1)的加标空白水样评估分析的准确度,平行重复测定五次,结果如表2所示。使用相对标准偏差(rsd)评估方法的精确度,所有目标分析物的相对标准偏差均在1.4-5.3%之间。因此,新开发的方法被认为是一种快速、高效、可靠,适用于水样中三唑类农药残留测定的方法。
表1
表2
为了评估方法,与文献已有报道的方法进行了一系列的比较,包括操作时间,样品基质,萃取剂,检测限,回收率等分析参数来评估。结果总结在表3中。与其他方法相比,本发明所提出的方法检测限相对较低,且操作时间明显低于其他方法,更为省时。此外,该方法使用少量的低共熔溶剂代替离子液体及其他高毒性的有机试剂作为萃取剂,减少了对环境的污染和对人体的伤害。即本发明所建立的方法是一种快速、简单、高效、可靠、环保的前处理方法,可以适用于不同体积样品的前处理操作,同时具有批量样品同时进行操作的优点。
表3
实施例2:环境水样中三唑类农药残留的检测(内标法)
方法同实施例1,不同之处在于低共熔溶剂为氯化胆碱(氢键受体):对氯苯酚(氢键供体)物质的量之比1:2的混合物。
制备:称取35g干燥过的氯化胆碱,64g苯酚,置于500ml圆底烧瓶中,并于80℃条件下持续搅拌2h直至形成澄清均一的液体溶剂。
最终获得水样中各三唑类农药的含量,结果如下:腈菌唑6.18μg·l-1、戊唑醇2.96μg·l-1、苯醚甲环唑4.48μg·l-1。
实施例3:环境水样中三唑类农药残留的检测(内标法)
方法同实施例1,不同之处在于低共熔溶剂为氯化胆碱(氢键受体):苯甲醇(氢键供体)物质的量之比1:3的混合物。
制备:称取35g干燥过的氯化胆碱,81g苯甲醇,置于500ml圆底烧瓶中,并于80℃条件下持续搅拌2h直至形成澄清均一的液体溶剂。
最终获得水样中各三唑类农药的含量,结果如下:腈菌唑6.19μg·l-1、戊唑醇3.12μg·l-1、苯醚甲环唑4.57μg·l-1。
- 还没有人留言评论。精彩留言会获得点赞!