一种测定玉米和小麦中痕量溴苯腈、碘苯腈和羟敌草腈残留量的气相色谱法的制作方法
本发明涉及有害痕量物质分析检测的方法,具体涉及到一种同时测定玉米和小麦中痕量溴苯腈、碘苯腈和羟敌草腈残留量的气相色谱法。
背景技术:
除草剂是农业生产中的重要物质,对于杀灭有害杂草、提高农产品产量起到了重大的作用,但是随着各类除草剂的广泛使用,除草剂滥用和残留问题也日益突出。溴苯腈、碘苯腈和羟敌草腈残留量是三种化学结构相似的除草剂,可用于防除阔叶杂草蓼、藜、苋、麦瓶草、龙葵、苍耳、麦家公、猪毛菜、田旋花、荞麦蔓等一年生双子叶杂草,广泛应用于玉米、小麦和高粱等农作物的生产领域中。毒理研究表明这三种物质具有中高毒性,对皮肤、眼有刺激作用,对鱼类低毒,对鸟类中等毒,同时具有污染池塘、河流和其他水源的风险。其中溴苯腈人体每日允许摄入量(adi)是0.05mg/kg,美国推荐的在谷物中的最高残留限量(mrl)为0.1mg/kg,要求严格。
当前关于食品中这类物质残留量的标准方法主要为《sn/t2228-2008进出口食品中31种酸性除草剂残留量的检测方法气相色谱-质谱法》,检测手段为气相色谱质谱联用法,前处理方法为凝胶渗透色谱法。应用凝胶渗透色谱法进行前处理可以去除大部分的杂质,起到较好的前处理效果,但是该前处理过程存在费时低效、需要耗费大量有机溶剂等诸多缺点,不适应大通量样品的快速筛查检测,亟待需要新的前处理方法来解决快速高效检测筛查的问题。
在前期的研究中发明人应用苯磺酸根-镁铝型水滑石焙烧产物吸附剂对水中的羟基苯甲腈类化工中间体进行了吸附性研究,取得了良好的选择吸附性效果。受此启发,发明人进一步应用苯磺酸根-镁铝型水滑石吸附剂和苯磺酸根-镁铝型水滑石焙烧产物吸附剂对溴苯腈、碘苯腈和羟敌草腈等三种除草剂进行吸附选择性研究,发现苯磺酸根-镁铝型水滑石吸附剂相较于其焙烧产物而言吸附此三种除草剂的效果更佳。在此基础上,发明人进一步对所研制的吸附剂富集三种目标化合物的性能和应用方法进行了优化,并根据三种目标物中含有卤素、可以微电子俘获检测器(μecd)上获得良好响应值的特点,建立了以苯磺酸根-镁铝型水滑石为吸附剂的玉米和小麦中痕量溴苯腈、碘苯腈和羟敌草腈残留量检测的气相色谱法。
技术实现要素:
为了克服现有标准方法中痕量溴苯腈、碘苯腈和羟敌草腈残留量检测前处理中使用凝胶渗透色谱费时低效、需要耗费大量有机溶剂、不适应于大通量样品筛查等诸不足,本发明要解决的技术问题在于提供一种基于新型吸附剂分散固相萃取快速吸附的、适合于玉米和小麦中痕量溴苯腈、碘苯腈和羟敌草腈残留量检测的气相色谱法。
本发明是通过以下技术方案来实现上述目标的。
一种测定玉米和小麦中痕量溴苯腈、碘苯腈和羟敌草腈残留量的气相色谱法,其特征在于,包括如下步骤:
步骤1化合物的提取和吸附:玉米和小麦样品粉碎后密封保存于4℃,使用时于具塞塑料离心管中称取样品5.00g,向离心管中添加碱性10%丙酮水溶液30ml,密闭并涡旋后超声提取,离心,将上清液转移至另一具塞塑料离心管中,重新提取一次并合并提取液;向离心管中添加0.5g苯磺酸根-镁铝型水滑石吸附剂,密闭并涡旋后震荡一定时间使吸附剂吸附提取液中的三种化合物;
步骤2化合物的脱吸附:将具塞离心管中离心,弃去上清液,向其中添加一定量的硫酸溶液溶解固体吸附剂,完成被吸附化合物的脱吸附;
步骤3化合物的萃取与衍生化:向上述离心管中添加一定量无水硫酸钠及有机溶剂萃取,涡旋、离心,分取上清液至衍生瓶中,向其中添加0.10ml乙酸酐和0.10g碳酸氢钠粉末,密闭并涡旋后置于温度为50℃的水浴中衍生化30min;
步骤4化合物的分析测试:将衍生瓶置于-10℃的冰箱中冷却10min至15min,向衍生瓶中添加2ml饱和碳酸氢钠水溶液,涡旋,静置分层,吸取下层溶液并向其中添加2.0g无水硫酸钠,涡旋,吸取上层有机溶液过滤后应用气相色谱法按照下列条件进行分析测试:
a)色谱柱:hp-5毛细管柱,30m×0.32mm,0.25μm膜厚;恒流模式,柱流速:1.00ml/min。
b)进样口温度:250℃;进样方式:不分流进样;进样量:2μl;尾吹气流量:60ml/min。
c)升温程序:60℃(保持2min),以10℃/min速率升温至150℃(保持1min),以5℃/min速率升温至180℃(保持2min),以20℃/min速率升温至250℃(保持0min),后运行300℃(保持3min)。
d)检测器:配备微电子俘获检测器,温度:325℃。
e)载气:高纯氮气(纯度≥99.999%)。
其中,
步骤1中所述的碱性10%丙酮水溶液为用0.01mol/l的氢氧化钠溶液调节ph=9.0,超声提取时间为20min,吸附震荡的时间为15min。
步骤2中所述的硫酸溶液为80%硫酸水溶液,用量为3.00ml。
步骤3中添加的无水硫酸钠为2.0g,有机萃取溶剂为5.00ml乙酸乙酯并全取用于衍生化反应。
步骤4中所述的滤膜为有机相滤膜,孔径0.22μm。
上述步骤中涡旋为涡旋1min至2min,离心为以4500rpm转速运行3min。
食品中溴苯腈、碘苯腈和羟敌草腈三种化合物可用丙酮、乙酸乙酯、乙腈等有机溶剂提取,同时这三个化合物具有一定的弱酸性,可溶解于碱性水溶液。在本发明的实验过程中发明人根据三个目标物的溶解性特征,结合目标可溶解于有机溶剂和碱性水溶液的特征,选择了可与水溶解的丙酮与水组成混合提取溶剂,并且调节了ph,以增加提取溶剂对目标物的提取效率,减少共提取杂质。
为了使吸附剂对目标物快速完全吸附,实验中根据样品量的大小优化了吸附剂的使用量;根据目标物可耐受较高浓度硫酸的特征,本发明中采用了80%的浓硫酸溶解吸附剂,并有效地借助80%浓硫酸可去除有机杂质的特征进一步地净化了共提取物杂质。发明人在实验中比较了甲醇-硫酸法、甲醇-氢氧化钠、三甲基硅烷化重氮甲烷法等多种酚类的衍生化方法,发现使用乙酸酐衍生化的方法对三种目标物的衍生化效果较好。
由于三种目标物中均含有卤素原子,在微电子俘获检测器上具有良好的响应值,因此本发明中采用了普及率更高的、配备微电子俘获检测器的气相色谱仪作为检测技术手段。
此外出于对目标物定量准确性考虑,本方法在无法获得目标物同位素从而进行同位素内标法定量的前提下采用基质校正曲线对目标物进行定量,以尽量消除系统误差提高定量的准确性。
本发明的优点在于:
(1)本发明所采用的新型吸附剂苯磺酸根-镁铝型水滑石可以采用分散固相萃取的方式对提取液中痕量溴苯腈、碘苯腈和羟敌草腈残留量进行快速的吸附,相较于标准方法的凝胶渗透色谱方式可以节省大量的吸附时间;
(2)本发明利用苯磺酸根-镁铝型水滑石吸附剂可溶解于酸的特点,应用盐酸溶液对吸附目标物后的吸附剂进行溶解,可以使得目标物从吸附剂上完全脱吸附,同时使用80%的硫酸具有一定氧化性能,可以去除部分的有机物杂质;
(3)本发明仅适用少量的有机溶剂作为目标物的萃取溶剂,相较于标准法方法需要使用大量有机溶剂具有显著的安全、环保优点和经济优势。
附图说明
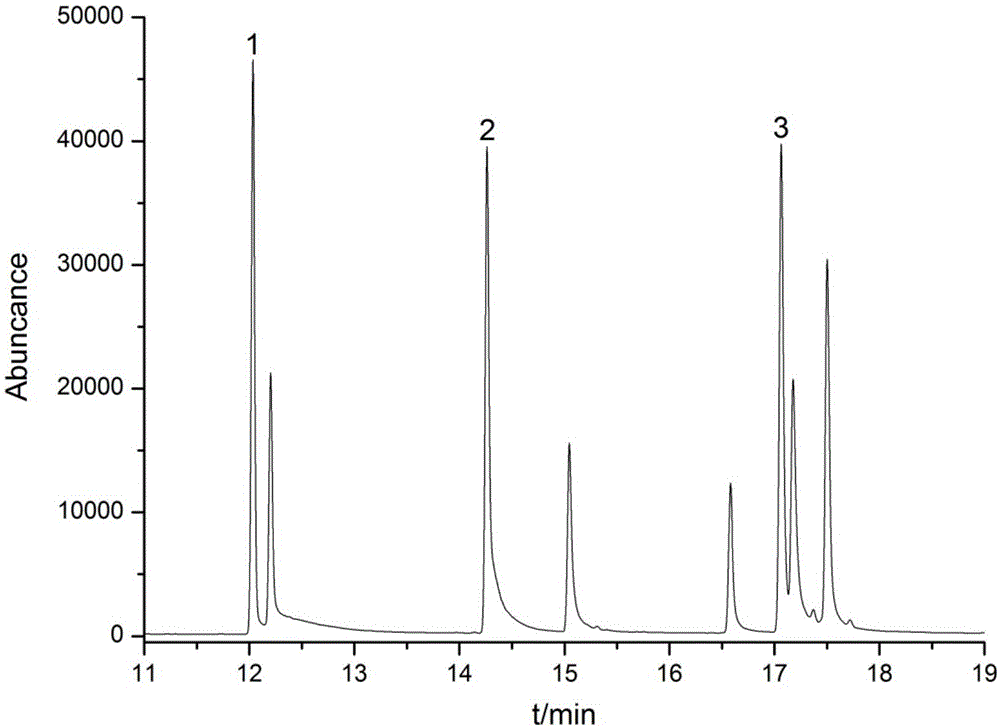
图1是具体实施方式中溴苯腈、碘苯腈和羟敌草腈残留量的基质标准溶液色谱图,浓度为200.0μg/kg,其中1是羟敌草腈、2是溴苯腈、3是碘苯腈。
具体实施方式
为进一步公开而不是限制本发明,以下结合实例对本发明作进一步的详细说明。
(1)本发明实施例中所涉及到的试剂药品如下:
溴苯腈、碘苯腈和羟敌草腈残留量共三种化合物固体标准品,纯度大于等于98.0%,上海阿拉丁生化科技股份有限公司;
乙酸酐,乙酸乙酯,丙酮,无水硫酸钠、氢氧化钠,碳酸氢钠,分析纯,国药集团;
硫酸,优级纯,国药集团;水为符合gb/t6682规定的一级水。
实验中所使用的玉米和小麦样品均购置福清当地超市,研磨并过60目筛,密封,4℃下保存,使用前取出回温至室温。
(2)本发明实施例中所涉及到的仪器如下:
kh-75a型电热恒温鼓风干燥箱,广州市康恒仪器有限公司;
7890b型气相色谱仪,配电子俘获检测器,美国安捷伦科技有限公司。
(3)气相色谱仪分析测试条件:
a)色谱柱:hp-5毛细管柱,30m×0.32mm,0.25μm膜厚;恒流模式,柱流速:1.00ml/min。
b)进样口温度:250℃;进样方式:不分流进样;进样量:2μl;尾吹气流量:60ml/min。
c)升温程序:60℃(保持2min),以10℃/min速率升温至150℃(保持1min),以5℃/min速率升温至180℃(保持2min),以20℃/min速率升温至250℃(保持0min),后运行300℃(保持3min)。
d)检测器:配备微电子俘获检测器,温度:325℃。
e)载气:高纯氮气(纯度≥99.999%)。
(4)基质校正曲线的制作和检出限、定量限的确定
准确称量上述溴苯腈、碘苯腈和羟敌草腈残留量并以甲醇溶解定容,配制成浓度为1000mg/l的标准储备液,-4℃下保存。使用时将标准储备液以去离子水逐步稀释,配制成浓度梯度为10.0μg/l、20.0μg/l、40.0μg/l、100.0μg/l和200.0μg/l的标准使用液。
在五个50ml具塞离心管中称取空白样品5.00g,分别添加上述标准使用液5.00ml,制备得到系列浓度的基质校正曲线,向离心管中添加ph=9的碱性10%丙酮水溶液30ml,密闭并涡旋后超声提取15min,离心,将上清液转移至另一100ml具塞塑料离心管中,重新提取一次并合并提取液;向离心管中添加0.5g苯磺酸根-镁铝型水滑石吸附剂,密闭并涡旋后震荡15min后使吸附剂吸附提取液中的目标化合物;
将上述具塞离心管中离心,弃去上清液,向其中添加3.00ml的80%硫酸溶液溶解固体吸附剂,完成被吸附化合物的脱吸附;
向上述离心管中添加2.0g无水硫酸钠及5.00ml乙酸乙酯萃取,涡旋、离心,分取上清液至衍生瓶中,向其中添加0.10ml乙酸酐和0.10g碳酸氢钠粉末,密闭并涡旋后置于温度为50℃的水浴中衍生化30min;
将衍生瓶置于-10℃的冰箱中冷却10min,向衍生瓶中添加2ml饱和碳酸氢钠水溶液,涡旋,静置分层,吸取下层溶液并向其中添加2.0g无水硫酸,涡旋,吸取上层有机溶液过孔径为0.22μm的有机相滤膜后应用气相色谱法进行分析测试。
以溴苯腈、碘苯腈和羟敌草腈在样品溶液的浓度为x轴,痕量溴苯腈、碘苯腈和羟敌草腈衍生物在气相色谱仪上的色谱峰峰面积为y轴绘制基质标准曲线并用于外标法定量。
以信噪比s/n的三倍值为方法的检测限(lod,lod=3s/n),以信噪比s/n的十倍为方法的定量限(loq,loq=10s/n),并结合添加基质的体积计算各个化合物在水中的检出限和定量限。
上述基质标准曲线相关参数、lod和loq相关信息见表1。
表1.痕量溴苯腈、碘苯腈和羟敌草腈残留量基质标准曲线、检测限和定量限相关信息
(5)苯磺酸根-镁铝型水滑石吸附剂的合成
为了是本领域的技术人员能够重复实现本发明的相关实验,现在提供一种本发明中使用的关键物质苯磺酸根-镁铝型水滑石吸附剂的合成方法,如下:
①合成吸附剂所涉及到的试剂药品如下:
苯磺酸钠,分析纯,上海韶远试剂有限公司;
mg6al2(oh)16co3·4h2o,分析纯,美国aldrich公司。
②合成吸附剂所涉及到的仪器如下:
excel型微波消解仪,上海屹尧仪器科技发展有限公司,消解罐体积为100ml;微波马弗炉(烧结炉),美国cem公司;vd53型真空干燥箱,德国宾德科技公司;hj-5多功能恒温搅拌器,金坛荣华仪器制造有限公司;fs-12型分液漏斗振荡器,日本新光科技有限公司;3k-15型离心机,德国sigma科技公司;bf518945c-1型箱式电阻炉(马弗炉),美国赛默飞世尔科技公司。
③合成吸附剂的具体步骤如下:
(a)焙烧:将购买的镁铝型水滑石mg6al2(oh)16co3·4h2o置于马弗炉中,以升温速率5℃/min加热至温度为500℃,焙烧6h,得到焙烧产物mg6al2o8(oh)2;
(b)称量:于微波消解罐中称取10.799g插层剂苯磺酸钠和7.236g的焙烧产物mg6al2o8(oh)2。
(c)微波晶化水热合成:将去离子水煮沸并保持30min,之后添加60ml至上述装有插层剂和焙烧产物的微波消解罐中,密闭,将微波消解罐置于微波消解仪中,在130~150℃下微波加热30min完成合成;
(d)洗涤与干燥:将微波罐中的全部固体与液体倒出,以煮沸30min以上的去除二氧化碳的去离子水经加热搅拌、振荡洗涤并离心,90℃下真空干燥12h,研磨保存。
实施例1
本实施例1采用玉米的空白样品基质进行加标回收实验以验证本发明方法的可行性,按照下列步骤进行处理:
1.化合物的提取吸附:
在50ml具塞离心管中称取空白样品5.00g,并向其中各添加5.00ml浓度分别为10.0μg/l、20.0μg/l和200.0μg/l的三种化合物标准溶液,制备得到三水平六平行的加标样品,向离心管中添加ph=9的碱性10%丙酮水溶液30ml,密闭并涡旋后超声提取15min,离心,将上清液转移至另一100ml具塞塑料离心管中,重新提取一次并合并提取液;向离心管中添加0.5g苯磺酸根-镁铝型水滑石吸附剂,密闭并涡旋后震荡15min后使吸附剂吸附提取液中的目标化合物;
2.化合物的脱吸附:
将上述具塞离心管中离心,弃去上清液,向其中添加3.00ml的80%硫酸溶液溶解固体吸附剂,完成被吸附化合物的脱吸附;
3.化合物的萃取与衍生化:
向上述离心管中添加2.0g无水硫酸钠及5.00ml乙酸乙酯萃取,涡旋、离心,分取上清液至衍生瓶中,向其中添加0.10ml乙酸酐和0.10g碳酸氢钠粉末,密闭并涡旋后置于温度为50℃的水浴中衍生化30min;
4、分析测试:
将衍生瓶置于-10℃的冰箱中冷却10min,向衍生瓶中添加2ml饱和碳酸氢钠水溶液,涡旋,静置分层,吸取下层溶液并向其中添加2.0g无水硫酸,涡旋,吸取上层有机溶液过孔径为0.22μm的有机相滤膜后应用气相色谱法进行分析测试。
本实施例1的加标回收率实验相关参数见表2。
表2玉米样品的添加浓度及回收率实验数据(n=6)
实施例2
本实施例2采用小麦的空白样品基质进行加标回收实验以验证本发明方法的可行性,按照下列步骤进行处理:
1.化合物的提取吸附:
在50ml具塞离心管中称取空白样品5.00g,并向其中各添加5.00ml浓度分别为10.0μg/l、20.0μg/l和200.0μg/l的三种化合物标准溶液,制备得到三水平六平行的加标样品,向离心管中添加ph=9的碱性10%丙酮水溶液30ml,密闭并涡旋后超声提取15min,离心,将上清液转移至另一100ml具塞塑料离心管中,重新提取一次并合并提取液;向离心管中添加0.5g苯磺酸根-镁铝型水滑石吸附剂,密闭并涡旋后震荡15min后使吸附剂吸附提取液中的目标化合物;
2.化合物的脱吸附:
将上述具塞离心管中离心,弃去上清液,向其中添加3.00ml的80%硫酸溶液溶解固体吸附剂,完成被吸附化合物的脱吸附;
3.化合物的萃取与衍生化:
向上述离心管中添加2.0g无水硫酸钠及5.00ml乙酸乙酯萃取,涡旋、离心,分取上清液至衍生瓶中,向其中添加0.10ml乙酸酐和0.10g碳酸氢钠粉末,密闭并涡旋后置于温度为50℃的水浴中衍生化30min;
4、分析测试:
将衍生瓶置于-10℃的冰箱中冷却10min,向衍生瓶中添加2ml饱和碳酸氢钠水溶液,涡旋,静置分层,吸取下层溶液并向其中添加2.0g无水硫酸,涡旋,吸取上层有机溶液过孔径为0.22μm的有机相滤膜后应用气相色谱法进行分析测试。
本实施例2的加标回收率实验相关参数见表3。
表3玉米样品的添加浓度及回收率实验数据(n=6)
以上所述实施例仅表达了本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员而言,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围,因此本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!