一种利用UPLC特征图谱鉴别百部药材基源的方法与流程
本发明属于药物分析及药物质量控制技术领域,具体涉及一种利用uplc特征图谱鉴别百部药材基源的方法。
背景技术:
百部为百部科植物直立百部(stemonasessilifoliamiq.)、蔓生百部(stemonajaponicamiq.)或对叶百部(stemonatuberosalour.)的干燥块根。春、秋二季采挖,除去须根,洗净,置沸水中略烫或蒸至无白心,取出,晒干。百部具有润肺下气止咳,杀虫的作用。为治疗肺痨之要药,既能抑制结核杆菌而杀痨虫,以绝其根本,又能化痰止咳,滋阴润肺,肺阴充足,化源有利,使肺恢复宣肃功能,主治百咳、久咳不止、肺结核等。据中国药典2015版收载,百部为常见多基源中药,来源于百部科植物直立百部stemonasessilifolia(miq.)miq.、蔓生百部stemonajaponica(bl.)miq.或对叶百部stemonatuberosalour.。查阅大量文献发现,百部因品种、产地不同,其主要化学成分的种类和含量有一定的差异。从结构类型的分布来看,对叶百部中以i型和ii型生物碱为主,直立百部中ii型生物碱较多,i型和iv型次之,蔓生百部中ii型和iv型生物碱较多。在生物活性研究方面,i、ii、iii型生物碱均有止咳作用,但以i型最好。从文献研究结果,对叶百部品种较优。
目前文献研究主要集中于百部药材或饮片的化学成分研究方面,而关于百部药材基源鉴别的研究较少。因此,有必要提供一种百部药材基源鉴别的方法。
技术实现要素:
为了克服上述现有技术的不足,本发明的首要目的在于提供一种利用uplc特征图谱鉴别百部药材基源的方法。
本发明是通过以下技术方案实现:
一种利用uplc特征图谱鉴别百部药材基源的方法,包括如下步骤:
(a)供试品溶液的制备:
分别取对叶百部药材和直立百部药材,研细,取0.25g,精密称定,置10ml量瓶中,加入50-100%甲醇适量,超声处理10-30分钟,取出,放冷,用50-100%甲醇定容至刻度,摇匀,滤过,即得;
(b)参照物溶液的制备:
取绿原酸对照品适量,加80%甲醇制成每1ml含绿原酸20μg,即得;再取对叶百部对照药材、直立百部对照药材0.5g,精密称定,置20ml量瓶中,加入80%甲醇适量,超声处理30min,取出,放冷,用80%甲醇定容至刻度,摇匀,用0.22um滤膜滤过,即得;
(c)色谱条件与系统适用性试验:
以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1%磷酸为流动相b,按下表进行梯度洗脱;流速:0.25ml/min;柱温为25℃;检测波长为210nm;
(d)特征图谱的建立:
分别精密吸取参照物溶液与供试品溶液各1μl,注入超高效液相色谱仪,测定,采用“中药色谱指纹图谱相似度评价系统”匹配,建立对叶百部药材和直立百部药材的特征图谱;
(e)鉴别依据:
将对叶百部药材和直立百部药材的特征图谱进行比较,对叶百部药材中确定具有8个特征峰,以峰3绿原酸峰为参照峰s峰,各特征峰的相对保留时间分别为:峰1:0.82、峰2:0.95、峰3:1.00、峰4:1.04、峰5:1.18、峰6:1.48、峰7:1.52、峰8:1.67,相对保留时间在规定值的±10%之内;直立百部药材中确定具有6个色谱峰,与对叶百部药材相比,缺少峰1和峰2,将峰1和峰2作为鉴别百部药材基源的依据;
(f)鉴别:
将待测百部样品与对叶百部与直立百部药材的特征图谱进行比较,确定百部药材基源;若待测百部样品色谱中具有峰1和锋2,则为对叶百部;若无峰1和锋2,则为直立百部。
作为本发明进一步优选的技术方案,所述利用uplc特征图谱鉴别百部药材基源的方法,还包括步骤(g)峰1和峰2的指认:采用uplc-q-tof/ms方法进行测定,
其中,液相条件为:以十八烷基硅烷键合硅胶为填充剂;流动相:a:0.1%磷酸;b:乙腈;柱温:30℃;样品室温度:10℃;流速:0.30ml/min;进样体积:2μl,负离子模式;流动相流动梯度见下表:
质谱条件:氮气作为质谱离子源的雾化、锥孔气;电喷雾负离子模式;毛细管电压:2.5kv;锥孔电压:40v;萃取锥孔电压:3v;离子源温度:100℃;脱溶剂气温度:350℃;反向锥孔气流:50l/h;脱溶剂气流速:600l/h;碰撞气流速:0.5ml/min;扫描时间:0.5s;扫描时间间隔:0.02s;质荷比范围:50–1200m/z;。
本发明通过特征峰的指认,所述峰1的归属为新绿原酸,峰2的归属为3-香豆酰奎宁酸。
本法通过对供试品溶液的制备条件进行了优化,对不同提取溶剂、不同提取方式、不同提取时间进行考察,优选的,步骤(a)供试品溶液的制备:分别取对叶百部药材和直立百部药材,研细,取0.25g,精密称定,置10ml量瓶中,加入80%甲醇适量,超声处理10分钟,取出,放冷,用80%甲醇定容至刻度,摇匀,滤过,即得。
作为本发明进一步优选的技术方案,所述的一种利用uplc特征图谱鉴别百部药材基源的方法,还包括步骤(h)绿原酸的含量测定:采用超高液相色谱仪测定,其中,
供试品溶液的制备为:
取百部药材,研细,取粉末0.25g,精密称定,置10ml量瓶中,加入70%乙醇,超声处理10分钟,取出,放冷,用70%乙醇定容至刻度,摇匀,滤过,取续滤液,即得;
色谱条件为:
以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1%磷酸为流动相b,按下表中的规定进行梯度洗脱;柱温为25℃以下;流速为0.25ml/min;检测波长为325nm;理论塔板数按绿原酸计算应不低于10000;
本发明与现有技术相比,具有如下有益效果:
本发明先建立百部药材的特征图谱,通过对比分析,确定了鉴别百部药材基源的依据,该方法简单,重现性良好,准确可靠,uplc的线性梯度洗脱的条件简单,便于操作,可用于百部药材基源的鉴别,为临床原料的控制提供了依据。
附图说明
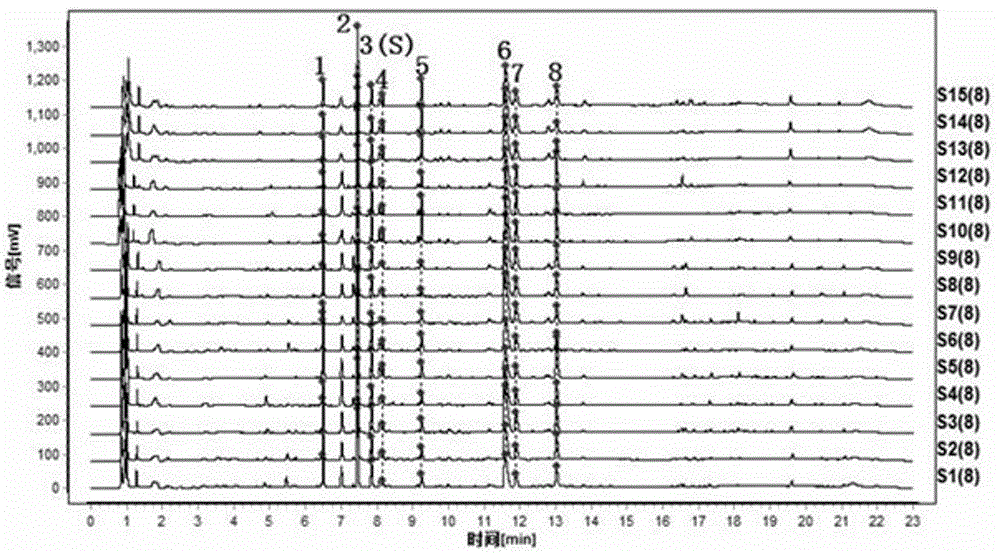
图1为15批对叶百部药材特征图谱的叠加图;
图2为对叶百部及直立百部药材基源鉴别图;
具体实施方式
下面通过具体实施方式来进一步说明本发明,以下实施例为本发明具体的实施方式,但本发明的实施方式并不受下述实施例的限制。
1、仪器与试药
仪器见表1、试剂见表2与试药见表3,药材信息见表4。
表1仪器信息汇总表
表2试剂信息汇总表
表3试药信息汇总表
表415批对叶百部药材产地信息
备注:s1-s6是野生来源。
2、色谱条件与供试品溶液的制备
2.1供试品溶液的制备
分别取对叶百部药材和直立百部药材,研细,取0.25g,精密称定,置10ml量瓶中,加入80%甲醇适量,超声处理10分钟,取出,放冷,用80%甲醇定容至刻度,摇匀,滤过,即得;2.2参照物溶液的制备
取绿原酸对照品适量,加80%甲醇制成每1ml含绿原酸20μg,即得;再分别取对叶百部对照药材、直立百部对照药材0.5g,精密称定,置20ml量瓶中,加入80%甲醇适量,超声处理(功率500w,频率40hkz)30min,取出,放冷,用80%甲醇定容至刻度,摇匀,用0.22um滤膜滤过,即得。
2.3色谱条件
以十八烷基硅烷键合硅胶为填充剂(柱长为10cm,内径为2.1mm,粒径为1.6μm;);以乙腈为流动相a,以0.1%磷酸为流动相b,按下表规定进行梯度洗脱;流速:0.25ml/min;柱温为25℃;检测波长为210nm。
2.4测定法
分别精密吸取对照品溶液与供试品溶液各1μl,注入超高液相色谱仪,测定。
3、供试品溶液制备方法考察
3.1提取溶媒考察
取对叶百部药材(批号:s1)粉末约0.25g,精密称定,分别加入80%甲醇、50%甲醇、甲醇、水和乙醇适量,超声处理(功率500w,频率40khz)10分钟,放冷,再用相应的溶剂定容至刻度,摇匀,离心(8000rpm,3min),上清液滤过,取续滤液,即得;按照“2.3”项下确定的色谱条件,进样,记录峰面积,结果如表5所示。
表5对叶百部药材特征图谱不同提取溶剂考察
结果表明,乙醇作为提取溶剂时色谱峰个数较少,水、甲醇、50%甲醇与80%甲醇的色谱峰个数及响应都相当,50%甲醇和80%甲醇作溶剂时的总峰面积/称样量较大,提取效率优于其余3种溶剂,80%甲醇的总特征峰面积/称样量值最大,综合考虑最终选择80%甲醇为提取溶剂。
3.2提取溶剂方式考察
取对叶百部药材(批号:s1)适量,研细,取约0.25g,精密称定,平行两份,一份置于10ml容量瓶中,加入80%甲醇适量,超声处理(功率500w,频率40khz)10分钟,放冷,用80%甲醇定容至刻度线;一份精密加入80%甲醇10ml,回流处理10分钟,放冷,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
结果显示,采用超声提取方式所得的色谱峰个数更多及响应更好,且操作简捷,故本方法选择超声处理作为供试液提取方式。
3.3提取时间考察
取对叶百部药材(批号:s1)适量,研细,取约0.25g,精密称定,平行三份,置10ml容量瓶中,加入80%甲醇适量,超声处理(功率500w,频率45khz)处理10分钟、20分钟、30分钟,放冷,分别用80%甲醇定容至刻度线,摇匀,离心(8000转,3分钟),滤过,取续滤液,即得;
结果表明,超声提取10分钟即可提取完全,为实验操作简便,最终选择超声处理10分钟。
4、色谱条件优化
4.1检测波长的确定
取对叶百部药材(批号:s1),研细,取约0.25g,精密称定,置10ml量瓶中,加入80%甲醇适量,超声(功率500w,频率40hkz)处理10min,取出,放冷,用80%甲醇定容至刻度,摇匀,用0.22um滤膜滤过,即得,记录190~400nm范围内的吸收光谱。
实验结果:通过移动波长,在210nm下,对叶百部药材样品溶液可以检测到的色谱峰信息最广,且各色谱峰的响应较大,且百部生物碱紫外吸收弱,多数在210nm吸收,因此选择210nm为检测波长。
4.2流动相的选择
取同一供试品溶液,分别考察流动相乙腈-0.002%三乙胺溶液、乙腈-0.1%磷酸、乙腈-0.1%甲酸为洗脱系统,按上述色谱条件进样分析。
实验结果,乙腈-0.002%三乙胺溶液作为流动相时,峰形较差,且基线不平稳;比较乙腈-0.1%磷酸和乙腈-0.1%甲酸,用0.1%甲酸在220nm等低波长干扰较大,用0.1%磷酸的色谱图较为理想。故选择乙腈-0.1%磷酸系统。
5、方法学考察
5.1专属性考察
精密吸取参照物溶液、供试品溶液、空白溶剂各1μl,注入液相色谱仪,按“2.3”项下色谱条件测定。结果显示,该分析方法能准确检测所指认的特征峰,不受提取溶剂的干扰,表明该色谱条件基本满足了信息量最大的原则。
5.2精密度考察
取对叶百部药材(s1),按供试品溶液制备方法制备成供试液,进样6次,每次1μl,考察特征峰相对保留时间和相对峰面积的一致性。结果显示:各特征峰相对保留时间的rsd最大的为0.51%,各峰相对保留时间及相对峰面积均在规定范围内,仪器精密度良好。
5.3重复性考察
取同一批号样品(批号:s1)共6份,按供试品溶液制备方法制备成供试液,进样1μl分析,考察特征峰相对保留时间和相对峰面积的一致性。结果显示:各特征峰相对保留时间最大的为0.75%,各峰相对保留时间及相对峰面积均在规定范围内,仪器重复性良好。
5.4稳定性考察
取同一批号样品(批号:s1),按供试品溶液制备方法制备成供试液,在0、2、4、6、8、12、16、20、24小时各进样一次,共测定24小时,分别进样1μl,考察特征峰相对保留时间和相对峰面积的一致性。结果显示:各特征峰的相对保留时间的rsd最大为0.69%,各峰相对保留时间及相对峰面积均在规定范围内,供试液在24小时内稳定性良好。
6、对叶百部药材特征图谱的建立
取对叶百部药材15批,按“2.1”项下制备成供试品溶液,按“2.3”项下的的色谱条件进样测定,测定结果见表6、7所示。将15批对叶药材特征图谱采用《中药色谱指纹图谱相似度评价软件系统》匹配,生成特征图谱叠加图,结果见图1。
表6样品测定结果(相对保留时间)
表7样品测定结果(相对峰面积)
实验结果表明:供试品特征图谱中应呈现8个特征峰,并应与对照药材参照物色谱中的8个特征峰相对应;与绿原酸参照物峰相应的峰为s峰,计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的±10%之内,规定值为:规定值为:0.82(峰1)、0.95(峰2)、1.04(峰4)、1.18(峰5)、1.48(峰6)、1.52(峰7)、1.67(峰8);计算各特征峰与峰5的相对峰面积,其相对峰面积应在规定的范围内,规定范围为:0.451~3.559(峰1)、1.579~7.511(峰2),0.420~2.649(峰3),0.137~0.726(峰4),1.808~16.747(峰6),0.421~4.460(峰7),0.420~3.957(峰8)。
7、鉴别依据:
按上述方法建立直立百部的特征图谱,直立百部药材中确定具有6个色谱峰,与对叶百部药材相比,缺少峰1和峰2(如图2),将峰1和峰2作为鉴别百部药材基源的依据;通过特征峰的指认,所述峰1的归属为新绿原酸,峰2的归属为3-香豆酰奎宁酸。
8、基源鉴别:
将待测百部样品与对叶百部与直立百部药材的特征图谱进行比较,确定百部药材基源;若待测百部样品色谱中具有峰1和锋2,则为对叶百部;若无峰1和锋2,则为直立百部。
9、峰1和锋2的指认:采用uplc-q-tof/ms方法进行测定:
9.1仪器与试剂
仪器:synaptg2hdms超高效液相飞行时间高分辨质谱联用系统(waterscorporation,milford,ma,usa),数据处理系统为markerlynx4.1工作站(waters,manchester,u.k.),ab135-s电子分析天平(梅特勒-托利多),kq-118b超声振荡仪(昆山市超声仪器有限公司)。
试剂:色谱乙腈和甲醇购于j.t.baker(phillipsburg,nj,usa),色谱级甲酸、磷酸、亮氨酸脑啡肽购于sigmaaldrich(mo,usa),实验超纯水(18.2mω)用milli-q水净化系统制备(millipore,france),其他所用试剂均为分析纯。
9.2实验方法
9.2.1供试品溶液的制备
取对叶百部药材,研细,取0.25g,精密称定,置10ml量瓶中,加入80%甲醇适量,超声处理10分钟,取出,放冷,用80%甲醇定容至刻度,摇匀,滤过,即得;
9.2.2分析测试条件
9.2.2.1液相条件:采用watersacquityuplc系统(pda检测器),配置二元溶剂输送系统和自动进样器模块。色谱柱:cortecsuplct3色谱柱(100mm×2.1mm,1.6μm);流动相:a:0.1%磷酸;b:乙腈;柱温:30℃;样品室温度:10℃;流速:0.30ml/min;进样体积:2μl;流动相流动梯度见下表:
9.2.2.2质谱条件:质谱采用waterssynaptg2hdms系统。氮气作为质谱离子源的雾化、锥孔气;电喷雾电离负离子模式;毛细管电压:2.5kv;锥孔电压:40v;萃取锥孔电压:3v;离子源温度:100℃;脱溶剂气温度:350℃;反向锥孔气流:50l/h;脱溶剂气流速:600l/h;碰撞气流速:0.5ml/min;扫描时间:0.5s;扫描时间间隔:0.02s;质荷比范围:50–1200m/z;数据采集形式:continuum;灵敏性:normal;动态范围:extended。
9.2.2.3实验结果:采用优化后的uplc-q-tof/ms分析方法,对对叶百部样本进行检测。通过化合物的色谱峰保留行为、精确分子量和mse碎片信息及标准品进行比对,对特征峰进行了指认。
峰1的鉴定:
负离子模式下,峰1在一级质谱图中给出m/z353.0898,为准分子离子峰[m-h]–(c16h17o9δm/z7.0ppm);在二级质谱中,产生了丰度较大的碎片离子峰m/z191,179和135;此外,该色谱峰的紫外光谱中存在240,280和320nm的特征峰。通过查阅文献比对,初步推断为新绿原酸,最后通过与标准品比对,确定其为新绿原酸(neochlorogenicacid)。
峰2的鉴定:
负离子模式下,峰2在一级质谱图中均给出m/z367.1049,为准分子离子峰[m-h]–(c17h19o9δm/z5.4ppm);二级质谱中,产生了碎片离子,分别为m/z193(100),191,173,134;此外,该色谱峰的紫外光谱中均存在240和320nm的特征峰。通过查阅文献比对,初步推断为阿魏酰奎宁酸类化合物。再根据其质谱的基峰离子,发现峰2碎片离子基峰为m/z193(100),推测其可能为3-阿魏酰奎宁酸(3-feruloylquinicacid)。
10、绿原酸含量测定
10.1、色谱条件与供试品溶液的制备
10.1.1供试品溶液的制备
取对叶百部药材适量,研细,取粉末0.25g,精密称定,置10ml量瓶中,加入70%乙醇适量,超声处理(500w,40khz)10分钟,取出,放冷,用70%乙醇定容至刻度,摇匀,滤过,取续滤液,即得。
10.1.2参照物溶液的制备
取绿原酸对照品适量,精密称定,加甲醇制成每1ml含绿原酸0.5mg的储备溶液;精密移取上述储备溶液1ml,用甲醇稀释成每1ml含绿原酸0.025mg的对照品溶液。
10.1.3色谱条件
色谱条件如下:以十八烷基硅烷键合硅胶为填充剂(柱长为10cm,内径为2.1mm,粒径为1.7μm);以乙腈为流动相a,以0.1%磷酸为流动相b,按下表中的规定进行梯度洗脱;柱温为25℃以下;流速为0.25ml/min;检测波长为325nm。理论塔板数按绿原酸计算应不低于10000。
10.1.4测定法
分别精密吸取对照品溶液与供试品溶液各1μl,注入超高液相色谱仪,测定。
10.2、方法学考察
10.2.1准确度
取已知含量对叶百部药材(批号:s5)约0.125g,精密称定,每批次平行称定6份,每份加入相近量的绿原酸对照品,按供试品溶液制备方法,每批次制备供试品溶液6份。每份精密吸取1μl按上述色谱条件测定,以下列公式计算回收率,rsd值,结果见表8。
表8加样回收率实验结果
实验结果显示对叶百部药材绿原酸的回收率均值为101.8%,rsd为2.2%,根据《中国药典》2015版四部“药品质量标准分析方法验证指导原则”规定样品中待测成分含量为0.1%时,回收率限度为90%~108%,表明回收率符合要求。
10.2.2精密度
精密吸取供试品溶液(批号:s5),按照上述色谱条件重复进样6次,进样体积1μl。计算峰面积rsd,rsd为0.21%,实验结果表明仪器精密度良好,测定结果见表9。
表9精密度考察结果
10.2.3重复性
取同一批供试品溶液6份(批号:s5),每份精密吸取1μl按上述色谱条件测定,测定供试品溶液中绿原酸含量,计算rsd,实验结果表明对叶百部药材含量结果rsd<3%,该分析方法的重复性良好,测定结果见表10。
表10重复性试验
10.2.4专属性
精密吸取供试品溶液(批号:s5)、绿原酸对照品溶液与空白溶剂各1μl,注入液相色谱仪,按照上述色谱条件以pda检测器进行210nm~400nm扫描检测。结果见表11-12。
表11色谱峰分离参数
表12色谱峰纯度参数
实验结果表明空白溶剂色谱中在与绿原酸相应的保留时间没有色谱峰,说明溶剂对绿原酸的测定无干扰;样品中绿原酸未检测到杂质峰,峰纯度因子在计算出的阈值限值内,说明在该色谱条件下,绿原酸峰纯度符合要求。综上所述,以本法测定绿原酸的含量具有专属性。
10.2.5线性
精密移取对照品储备液适量,分别用甲醇稀释成系列浓度的对照品溶液,摇匀,按上述色谱条件依次进样1μl,记录峰面积,结果如表13:
表13绿原酸线性考察结果
线性回归方程为:y=12496342.3453x-1903.8887,相关系数r2=1.0000,表明绿原酸在5.47μg/ml~43.74μg/ml浓度范围内线性关系良好。
10.3绿原酸含量测定
将15批对叶百部药材按照上述方法制备成供试品溶液,测定含量,结果见表14。
表1415批对叶百部药材的绿原酸含量结果
- 还没有人留言评论。精彩留言会获得点赞!