利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法与流程
本发明涉及纳米材料、全息光栅和拉曼光谱领域,具体涉及一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法。
背景技术:
光致聚合物是数字全息存储最有希望最先得到实用的材料,为了改善其全息记录性能,人们通常在其中掺入无机纳米粒子,因此对掺入纳米粒子后材料记录过程中光化动力学机制的掌握有利于优化材料的化学成分和数字全息记录性能。然而,人们对掺纳米粒子的光致聚合物的光化动力学过程涉及到的光物理和光化学机制理解不够深入,在光致聚合物体系中纳米粒子常常会团聚,且在光致聚合的过程中无法避免纳米粒子与光敏剂之间的电荷转移,进而影响单体聚合的过程。此外,很少有人去研究纳米粒子的扩散和单体扩散的问题,对这种体系,多组分扩散动力学问题的研究更少。问题的关键是人们缺少有效的方法去研究纳米粒子在光致聚合物种的分布情况,从而限制了人们更深入理解光化动力学过程,因此亟待开发一种检测光致聚合物中纳米粒子空间分布的方法。
表面增强拉曼光谱是一种基于表面等离激元共振的纳米光子学技术,因为能够提供类似于“指纹”一样的特征振动光谱信息,而在材料科学、表面科学、生命科学以及人们实际生活中备受关注。拉曼图像是描绘样品上面或里面不同点的光谱信息的改变,它可以以一维轮廓、二维图像或三维着色体积的形式存在,可以通过拉曼成像结果显示随位置改变的拉曼光谱。
技术实现要素:
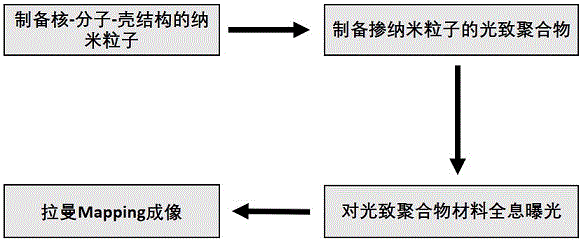
本发明提出了一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法,通过拉曼光谱仪器逐点扫描成像采集光致聚合物干涉区域同一直线上不同位置的拉曼光谱,根据拉曼mapping图纳米粒子的分布情况,即可知道光致聚合物经过全息记录后纳米粒子的分布情况。
实现本发明的技术方案是:
一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法,将纳米复合材料加入到光致聚合物体系中并进行全息曝光,利用拉曼光谱仪进行mapping成像测试,获得拉曼成像图,得到纳米粒子的空间位置分布情况。
所述纳米复合材料首先合成金纳米粒子,合成的金纳米粒子的表面吸附一层对巯基苯甲酸分子并包覆极薄且致密的二氧化硅壳层,形成为以金为核层,二氧化硅为壳层,对巯基苯甲酸分子层在中间的au-mba-sio2纳米粒子(简称cms结构)。
所述核-分子-壳纳米复合材料的壳层厚度为2~3nm,对巯基苯甲酸分子与金纳米粒子通过au-s化学键结合,核层金的尺寸为15~20nm。
纳米复合材料(cms纳米粒子)的具体制备步骤如下:
(1)将对巯基苯甲酸溶液加入到金胶溶液中搅拌至混合均匀后,60~70℃保温12~18h完成金纳米粒子表面对巯基苯甲酸分子的吸附;
(2)将步骤(1)吸附完对巯基苯甲酸分子的金纳米粒子溶胶加入到3-氨丙基三甲氧基硅烷水溶液中,搅拌10~15min,然后加入一定量的硅酸钠水溶液,搅拌3~5min;然后将反应容器置于90~95℃的水浴中加热反应1~2h,在冰水中终止反应完成au-mba-sio2纳米复合材料的合成。
所述步骤(1)中对巯基苯甲酸溶液的浓度为20~25mm,对巯基苯甲酸溶液和金胶溶液的体积比为1:(100~200)。
所述步骤(2)中3-氨丙基三甲氧基硅烷水溶液的浓度为1~3mmlo/l,硅酸钠水溶液的质量浓度为0.38~0.54%,以步骤(1)中对巯基苯甲酸溶液的体积为基准,3-氨丙基三甲氧基硅烷水溶液的加入量为0.4~0.6ml,硅酸钠水溶液的加入量为3.0~3.5ml。
所述光致聚合物为28~36wt%的聚乙烯醇、9~14wt%的丙烯酰胺、22~25wt%的三乙醇胺和30~34wt%的亚甲基蓝制备得到。
所述光致聚合物的制备方法如下:
(1)在温度为25-28℃,相对湿度为45%~60%的暗室里,将聚乙烯醇溶于超纯水中加热80℃搅拌直至聚乙烯醇完全溶解;
(2)将单体丙烯酰胺溶于超纯水中,并且加热至30℃使其完全溶解后加入到步骤(1)的聚乙烯醇溶液中;
(3)将三乙醇胺加入到步骤(2)混合溶液中,混合后加入浓度为2×10-3~3.5×10-4m的亚甲基蓝溶液,搅拌均匀得到光致聚合物。
利用拉曼光谱仪进行成像测试,所述的光路采用的是对称入射光路非倾斜光栅对掺纳米粒子的光致聚合物进行全息曝光,其物光和参考光的夹角θ=5~15度,全息光栅的条纹间距大于激光拉曼光谱仪激光光斑的直径。光致聚合物经过全息记录后,用激光拉曼光谱仪进行mapping测试可获得分子的拉曼信号,根据拉曼mapping图即可知道粒子的分布情况。
本发明的有益效果是:
(1)本发明所合成的纳米粒子是以金为核,二氧化硅为壳,分子层在中间的核-分子-壳纳米材料。其中金核显著增强了中间层分子的拉曼信号,二氧化硅壳能隔绝金核和光敏剂分子之间的电荷传输,增强了光致聚合物材料的稳定性。
(2)本发明只需将掺有cms的纳米粒子光致聚合物进行全息曝光,用拉曼光谱仪mapping模式下对对巯基苯甲酸分子的信号进行线扫描采集信号,然后mapping成像即可获得粒子的分布情况。
(3)本发明解决了在全息存储中,人们不清楚掺纳米粒子的光致聚合物经全息曝光后粒子的分布情况,合成的核-分子-壳结构的纳米粒子有效隔绝了光敏剂与纳米粒子之间发生的电荷转移,有助于帮助人们更好地理解全息动力学。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的流程图。
图2为制备核-分子-壳的实验流程示意图。
图3和4分别为实施例1和实施例2中金纳米粒子及au-mba-sio2纳米粒子的吸收光谱。
图5和6分别为实施例1和实施例2中金纳米粒子的透射电子显微镜图(tem)。
图7和8分别为实施例1和实施例2中au-mba-sio2纳米粒子的透射电子显微镜图。
图9为实施例2对光致聚合物全息记录所用的光路图。
图10和11为实施例1检测全息曝光后干涉区域内样品在同一直线上不同位置cms结构内标分子mba1585cm-1处的特征峰峰强与位置的关系图及对应的mapping图。
图12和13为实施例2检测全息曝光后干涉区域内样品在同一直线上不同位置cms结构内标分子mba1585cm-1处的特征峰峰强与位置的关系图及对应的mapping图。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法,步骤如下:
(1)au-mba-sio2纳米粒子的制备:
取240ul浓度为20mm的对巯基苯甲酸溶液加入到30ml新鲜的平均粒径约为15±3nm金胶溶液中搅拌至混合均匀后,60度保温12h即可完成金纳米粒子表面对巯基苯甲酸分子的吸附过程。然后,将溶液置于圆底烧瓶中,加入0.4ml1mmol/l的3-氨丙基三甲氧基硅烷水溶液并搅拌10min。再加入3.0ml浓度为0.54%的硅酸钠水溶液继续搅拌5min后。然后将反应溶液置于90度的水浴中加热反应1h,然后终止反应冷却至室温便可制得壳厚为2nm的au-mba-sio2纳米粒子。
核-分子-壳结构的合成示意图如附图2所示,图3、5和7为金纳米粒子及au-mba-sio2纳米粒子的吸收光谱和透射电子显微镜图(tem)。
(2)全息记录样品的制备
在温度为25°c,相对湿度为45%的暗室里,将1.7g聚乙烯醇(pva)溶于17.5ml的超纯水中加热80度搅拌直至pva完全溶解。将0.8g的单体丙烯酰胺(aa)溶于一定的超纯水中,并且加热至30度使其完全溶解后加入到pva溶液中。取1.5ml三乙醇胺(tea)加入到上述溶液中后再加入2ml浓度为2×10-3m的亚甲基蓝溶液中搅拌均匀,即可得到聚合物溶液。
(3)取适量的光致聚合物溶液与纳米粒子混合均匀后,取适量的溶液滴涂到干净的2cm×2cm玻璃基片上面,在暗室中干燥36h即可形成掺纳米粒子光致聚合物干膜,下表为掺纳米粒子光致聚合物材料的基本组成。
掺纳米粒子光致聚合物材料的基本组成
光致聚合物的全息记录过程
采用附图9所示的光路,参考光和物光对称入射相交入射到样品上,两束光的夹角为5°,记录为非倾斜光栅。激光器为633nmhe-ne激光器,物光和参考光的光强比值为1:1,两束光的记录光强为15mw/cm2,记录时间为60s。
用拉曼光谱仪对曝光后的光致聚合物mapping测试。
因为光敏剂亚甲基蓝对785nm波段几乎没有吸收,所以采用785nm的激光光源采集样品的拉曼信号。mapping模式下进行测试时的步长为1um,线扫描总步长30um,衰减功率0.1%,积分时间1s,选取探针分子mba1585cm-1处的峰强度进行拟合,测试结果如图10和11所示。通过图10及11可以观察到1585cm-1处的峰强呈现周期性分布,因此我们可以得知样品经过全息记录后,纳米粒子的分布是呈现周期性分布。这一结果清楚地告诉了人们在全息存储中掺杂纳米粒子的光致聚合物经过全息曝光后,粒子是呈现周期性规律分布的,除了形成光产物光栅外,还会形成纳米粒子吸收性光栅。这一检测方法,有助于帮助人们更好地理解全息动力学过程。
实施例2
一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法,步骤如下:
(1)au-mba-sio2纳米粒子的制备:
取240ul浓度为25mm的对巯基苯甲酸溶液加入到30ml新鲜的平均粒径约为20±3nm金胶溶液中搅拌至混合均匀后,65度保温18h即可完成金纳米粒子表面对巯基苯甲酸分子的吸附过程。然后,将溶液置于圆底烧瓶中,加入0.6ml1.5mmol/l的3-氨丙基三甲氧基硅烷水溶液并搅拌15min。再加入3.5ml浓度为0.45%的硅酸钠水溶液继续搅拌10min后。然后将反应溶液置于95度的水浴中加热反应1.5h,然后终止反应冷却至室温便可制得壳厚为3nm的au-mba-sio2纳米粒子。
核-分子-壳结构的合成示意图如附图2所示,图4、6和8为金纳米粒子及au-mba-sio2纳米粒子的吸收光谱和透射电子显微镜图(tem)。
(2)全息记录样品的制备
在温度为28°c,相对湿度为60%的暗室里,将2.3g聚乙烯醇(pva)溶于17.5ml的超纯水中加热85度搅拌直至pva完全溶解。将0.6g的单体丙烯酰胺(aa)溶于一定的超纯水中,并且加热至30度使其完全溶解后加入到pva溶液中。取1.5ml三乙醇胺(tea)加入到上述溶液中后再加入2ml浓度为3.5×10-4m的亚甲基蓝溶液中搅拌均匀,即可得到聚合物溶液。
(3)取适量的光致聚合物溶液与纳米粒子混合均匀后,取适量的溶液滴涂到干净的2cm×2cm玻璃基片上面,在暗室中干燥48h即可形成掺纳米粒子光致聚合物干膜,下表为掺纳米粒子光致聚合物材料的基本组成。
掺纳米粒子光致聚合物材料的基本组成
光致聚合物的全息记录过程
采用附图9所示的光路,参考光和物光对称入射相交入射到样品上,两束光的夹角为10°,记录为非倾斜光栅。激光器为633nmhe-ne激光器,物光和参考光的光强比值为1:1,两束光的记录光强为15mw/cm2,记录时间为85s。
用拉曼光谱仪对曝光后的光致聚合物mapping测试。
因为光敏剂亚甲基蓝对785nm波段几乎没有吸收,所以采用785nm的激光光源采集样品的拉曼信号。mapping模式下进行测试时的步长为0.8um,线扫描总步长36um,衰减功率0.1%,积分时间1s,选取探针分子mba1585cm-1处的峰强度进行拟合,测试结果如图12和13所示。通过图12及13可以观察到1585cm-1处的峰强呈现周期性分布,因此我们可以得知样品经过全息记录后,纳米粒子的分布是呈现周期性分布。这一结果清楚地告诉了人们在全息存储中掺杂纳米粒子的光致聚合物经过全息曝光后,粒子是呈现周期性规律分布的,除了形成光产物光栅外,还会形成纳米粒子吸收性光栅。这一检测方法,有助于帮助人们更好地理解全息动力学过程。
实施例3
一种利用拉曼成像技术检测光致聚合物中纳米粒子空间分布的方法,步骤如下:
(1)au-mba-sio2纳米粒子的制备:
取300ul浓度为23mm的对巯基苯甲酸溶液加入到30ml新鲜的平均粒径约为20±3nm金胶溶液中搅拌至混合均匀后,70度保温16h即可完成金纳米粒子表面对巯基苯甲酸分子的吸附过程。然后,将溶液置于圆底烧瓶中,加入0.6ml3mmol/l的3-氨丙基三甲氧基硅烷水溶液并搅拌15min。再加入3.2ml浓度为0.38%的硅酸钠水溶液继续搅拌10min后。然后将反应溶液置于95度的水浴中加热反应2h,然后终止反应冷却至室温便可制得壳厚为3nm的au-mba-sio2纳米粒子。
(2)全息记录样品的制备
在温度为28°c,相对湿度为60%的暗室里,将2g聚乙烯醇(pva)溶于17.5ml的超纯水中加热85度搅拌直至pva完全溶解。将0.6g的单体丙烯酰胺(aa)溶于一定的超纯水中,并且加热至30度使其完全溶解后加入到pva溶液中。取1.5ml三乙醇胺(tea)加入到上述溶液中后再加入2ml浓度为3.5×10-4m的亚甲基蓝溶液中搅拌均匀,即可得到聚合物溶液。
(3)取适量的光致聚合物溶液与纳米粒子混合均匀后,取适量的溶液滴涂到干净的2cm×2cm玻璃基片上面,在暗室中干燥48h即可形成掺纳米粒子光致聚合物干膜,下表为掺纳米粒子光致聚合物材料的基本组成。
掺纳米粒子光致聚合物材料的基本组成
光致聚合物的全息记录过程
采用附图9所示的光路,参考光和物光对称入射相交入射到样品上,两束光的夹角为15°,记录为非倾斜光栅。激光器为633nmhe-ne激光器,物光和参考光的光强比值为1:1,两束光的记录光强为25mw/cm2,记录时间为45s。
用拉曼光谱仪对曝光后的光致聚合物mapping测试。
因为光敏剂亚甲基蓝对785nm波段几乎没有吸收,所以采用785nm的激光光源采集样品的拉曼信号。mapping模式下进行测试时的步长为1.5um,线扫描总步长40um,衰减功率0.1%,积分时间1s,选取探针分子mba1585cm-1处的峰强度进行拟合。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!