一种莫匹罗星软膏杂质定性定位、测试方法及应用与流程
本发明属于药物杂质检验领域,具体涉及一种莫匹罗星软膏杂质定性定位、测试方法及应用。
背景技术:
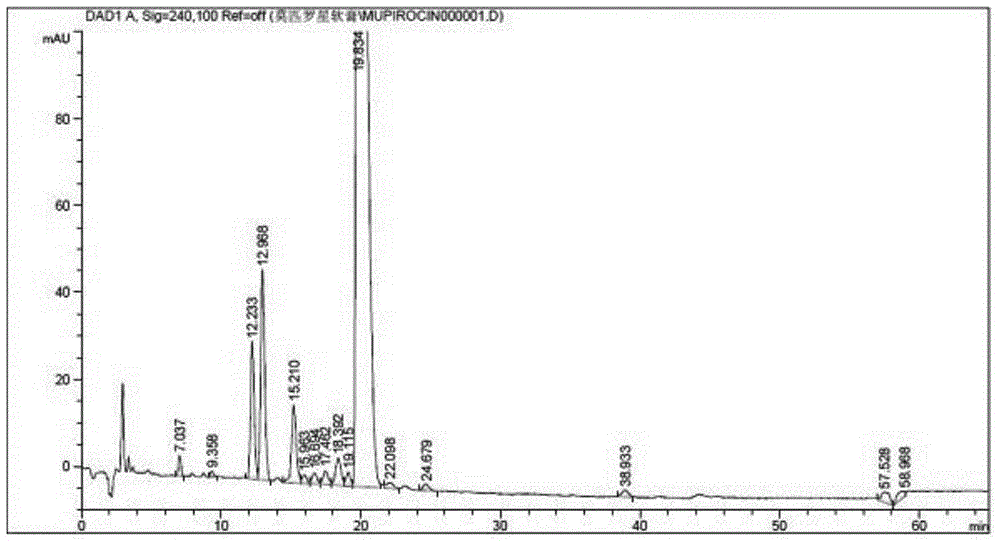
:莫匹罗星,又名假单胞菌酸a,是于1971年从荧光假单胞杆菌中分离得到的一种细菌代谢产物,主要用于预防和治疗革兰阳性致病菌,尤其是金黄色葡萄球菌,为类白色固体,由荧光假单孢菌浸没发酵后产生。由荧光假单孢菌产生的代谢产物包括有假单孢菌酸a、b、c、d、e、f。假单孢酸a,代表其活性的主要部分,占90~95%,其他代谢产物无生物活性。包括甲氧西林耐药型金黄色葡萄球菌以及链球菌造成的皮肤细菌感染,药剂主要为软膏,是皮肤科局部抗菌治疗的首选药物。《中国药典》2010年版尚未收载莫匹罗星软膏,但《国家药品标准》新药转正标准第23册、美国药典35版(usp35)、英国药典2012年版(bp2012)及日本药局16版(jp16)中均收载了莫匹罗星软膏(jp16收载为莫匹罗星钙软膏)。bp2009版莫匹罗星软膏中也将这些无生物活性的代谢产物作为杂质控制,控制了3个已知杂质,总杂质不得过20%。2011年5月出版的usp34版莫匹罗星软膏中控制了9个已知杂质,总杂质不得过30%。在对莫匹罗星软膏杂质进行检测时,往往需要购买杂质标准品,然后对其进行色谱检测后进行比对,并不是所有的杂质标准品易于得到,为更好地控制杂质含量,需要一种新的杂质定量方法确保简便、精密且全面的控制杂质。技术实现要素:为解决现有技术中在研制莫匹罗星软膏新药时,其杂质检测难以获得标准比对的技术问题,本发明提供一种莫匹罗星软膏杂质定性定位、测试方法及应用。一种莫匹罗星软膏杂质定性定位方法,其不同之处在于,包括如下步骤:步骤s1:将莫匹罗星软膏样品制备成定位测试样品;步骤s2:对所述测试样品进行液-质联动进行检测;步骤s3:对检测结果进行分析,确定各杂质结构及各杂质相对保留时间,得到杂质标准质谱图与杂质标准色谱图;其中,所述步骤s2中,色谱检测条件为:采用c8色谱柱,柱温为33℃~37℃,检测波长为240nm,流速:1ml/min,进样体积:19μl~21μl,采用流动相a及流动相b进行梯度洗脱;所述流动相a由质量百分比为75%的0.1mol/l醋酸铵及质量百分比为25%四氢呋喃的组成,所述流动相b由质量百分比为70%的0.1mol/l醋酸铵及质量百分比为30%的四氢呋喃组成;所述步骤s2中,质谱检测条件为:一级质谱:采用esi源,scan模式采集数据,扫描离子采用负模式,frag电压为135v,质量数扫描范围为:300-700;二级质谱:母离子选择500.3,frag电压为135v,碰撞电压20mv,production扫描范围为:100-500。进一步,所述步骤s1中,将所述莫匹罗星软膏样品静置后,称取莫皮软膏样品后依次加入四氢呋喃及醋酸钠后进行微孔过滤,将滤液收集得到所述定位测试样品。与现有技术相比,本发明定性定位方法是将研制样品直接制备成定位测试样品进行液-质联动检测得到,测试条件合理,可以控制莫匹罗星的九种杂质,可以直接成为后续杂质含量的参照,而无需购买杂质参照品进行比对。进一步,所述步骤s2中,色谱检测条件为:采用zorbaxeclipsexdb-c8色谱柱,柱温为35℃,检测波长为240nm,流速:1ml/min,进样体积:20μl。进一步,所述步骤s2中,梯度洗涤的条件参数为:时间(min)流动相a(%)流动相b(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度一种莫匹罗星软膏杂质测试方法,其不同之处在于,所述莫匹罗星软膏杂质测试方法包括以下步骤:步骤a1:将莫匹罗星软膏样品制备成含量测试样品;步骤a2:将所述含量测试样品进行高效色相色谱检测;步骤a3:将所述步骤a2的结果与杂质标准色谱图进行比对,得到所述莫匹罗星软膏杂质含量;其中,所述杂质标准色谱通过上述莫匹罗星软膏杂质定性定位方法检测得到。进一步,所述莫匹罗星软膏的包括以下重量百分比的成分:聚乙二醇-400:78.4%,聚乙二醇-4000:19.6%及莫匹罗星:2%。进一步,所述步骤a2中,采用c8色谱柱,柱温为33℃~37℃,检测波长为240nm,流速:1ml/min,进样体积:20μl,采用流动相c及流动相d进行梯度洗脱;所述流动相c及所述流动相d均0.1mol/l醋酸铵及四氢呋喃组成;其中,所述流动相c中,0.1mol/l醋酸铵的质量百分比为74%~76%,四氢呋喃的质量百分比为24%~26%;所述所述流动相d中,0.1mol/l醋酸铵的质量百分比为69%~71%,四氢呋喃的质量百分比为29%~31%。进一步,所述步骤a2中,采用c8色谱柱,柱温为35℃,检测波长为240nm,流速:1ml/min,进样体积:20μl,采用流动相c及流动相d进行梯度洗脱;所述流动相c及所述流动相d均0.1mol/l醋酸铵及四氢呋喃组成;其中,所述流动相c中,0.1mol/l醋酸铵的质量百分比为75%,四氢呋喃的质量百分比为25%;所述所述流动相d中,0.1mol/l醋酸铵的质量百分比为70%,四氢呋喃的质量百分比为30%。进一步,所述步骤a2中,采用zorbaxeclipsexdb-c8色谱柱或zorbaxeclipseplusc8色谱柱进行检测。进一步,所述步骤a2中,梯度洗涤的条件参数为:时间(min)流动相c(%)流动相d(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度与现有技术相比,本发明检测方法的有益效果为:在检测杂质含量时直接与产品本身的标准色谱进行比对,并结合合理的色谱测试条件,无需购买标准品再次检测比对,操作更加快速,结果更加精密可靠。附图说明图1为定位测试样品典型色谱图;图2为莫匹罗星软膏全扫描图谱;图3为rrt0.36色谱峰对应一级质谱;图4为rrt0.62色谱峰对应一级质谱;图5为rrt0.65色谱峰对应一级质谱;图6为rrt0.77色谱峰对应一级质谱;图7为rrt0.93色谱峰对应一级质谱;图8为rrt1.12色谱峰对应一级质谱;图9为rrt1.24色谱峰对应一级质谱;图10为rrt1.96色谱峰对应一级质谱;图11为rrt2.13色谱峰对应一级质谱;图12为莫匹罗星二级质谱图;图13为rrt0.62色谱峰对应二级质谱;图14为rrt0.65色谱峰对应二级质谱;图15为rrt1.12色谱峰对应二级质谱;图16为rrt1.24色谱峰对应二级质谱;图17波长=240nm含量测试样品典型色谱图;图18波长=240nm莫匹罗星对照品溶液典型色谱图图19含量测试样品各杂质紫外吸收光谱图;图20含量对照品溶液莫匹罗星紫外吸收光谱图;图21波长=220nm含量测试样品典型色谱图;图22波长=230nm含量测试样品典型色谱图;图23波长=240nm含量测试样品典型色谱图。具体实施方式以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。本发明实施例对下面重量百分比的新型莫匹罗星软膏进行测试:聚乙二醇-400:78.4%,聚乙二醇-4000:19.6%及莫匹罗星:2%。实施例一杂质的定性定位1.1仪器agilent1200高效液相色谱系统;agilentlc-ms/ms6460,esi源。1.2色谱条件及质谱条件1.2.1色谱条件色谱柱:zorbaxeclipsexdb-c8(250mm×4.6mm,5μm)检测波长:240nm流速:1ml/min进样体积:20μl柱温:35℃流动相:溶液a:0.1m醋酸铵∶四氢呋喃=75∶25溶液b:0.1m醋酸铵∶四氢呋喃=70∶30梯度表时间(min)流动相a(%)流动相b(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度1.2.2质谱条件:一级质谱:采用esi源,scan模式采集数据,扫描离子采用负模式,frag电压为135v,质量数扫描范围为:300-700。二级质谱:鉴于分子量为500.3的物质有多种,进行二级质谱扫描确定具体结构。母离子选择500.3,frag电压为135v,碰撞电压20mv,production扫描范围为:100-500。1.3定位测试样品制备新型莫匹罗星软膏样品置100℃环境中放置48小时,取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为定位测试样品(每1ml含约莫匹罗星5mg)。1.4精密量取供试品溶液20μl,注入液相色谱仪,记录色谱图及质谱图,典型色谱图及质谱图及定位测试样品全扫描色谱图如下:1.5针对定位测试样品全扫描色谱图进行结构解析一级质谱分析rrt0.36色谱峰,测得的[m-h]-为471.3,分子量为472.3。对照usp34中的杂质为假单胞酸f,化学式为c24h40o9,化学名称为:7-((e)-4-((2s,3r,4r,5s)-3,4-dihydroxy-5-(((2s,3s)-3-((2s,3s)-3-hydroxybutan-2-yl)oxiran-2-yl)methyl)tetrahydro-2h-pyran-2-yl)-3-methylbut-2-enoyloxy)heptanoicacid。对应一级质谱图3所示,对应结构式如式i所示。rrt0.62色谱峰,测得的[m-h]-为499.3,分子量为500.3。其分子量与usp34中相对应的莫匹罗星杂质1、莫匹罗星杂质2、莫匹罗星、莫匹罗星杂质3和莫匹罗星杂质4的分子量相同。需要进一步做二级质谱研究。对应一级质谱图4。rrt0.65色谱峰,测得的[m-h]-为499.3,分子量为500.3。其分子量与usp34中相对应的莫匹罗星杂质1、莫匹罗星杂质2、莫匹罗星、莫匹罗星杂质3和莫匹罗星杂质4的分子量相同。需要进一步做二级质谱研究。对应一级质谱图5。rrt0.77色谱峰,测得的[m-h]-为497.3,分子量为498.3。对照usp34中的杂质为假单胞酸d,化学式为c26h42o9,化学名称为:(e)-9-((e)-4-((2s,3r,4r,5s)-3,4-dihydroxy-5-(((2s,3s)-3-((2s,3s)-3-hydroxybutan-2-yl)oxiran-2-yl)methyl)tetrahydro-2h-pyran-2-yl)-3-methylbut-2-enoyloxy)non-4-enoicacid。对应一级质谱图6所示,对应结构式如式ii所示。rrt0.93色谱峰,测得的[m-h]-为515.3,分子量为516.3。对照usp34中的杂质为假单胞酸b,化学式为c26h44o10,化学名称为:9-((e)-3-methyl-4-((2s,3r,4s,5r)-3,4,5-trihydroxy-5-(((2s,3s)-3-((2s,3s)-3-hydroxybutan-2-yl)oxiran-2-yl)methyl)tetrahydro-2h-pyran-2-yl)but-2-enoyloxy)noninoicacid。对应一级质谱图如7,结构式如式iii所示。rrt1.12色谱峰,测得的[m-h]-为499.3,分子量为500.3。其分子量与usp34中相对应的莫匹罗星杂质1、莫匹罗星杂质2、莫匹罗星、莫匹罗星杂质3和莫匹罗星杂质4的分子量相同。需要进一步做二级质谱研究。对应一级质谱图8所示。rrt1.24色谱峰,测得的[m-h]-为499.3,分子量为500.3。其分子量与usp34中相对应的莫匹罗星杂质1、莫匹罗星杂质2、莫匹罗星、莫匹罗星杂质3和莫匹罗星杂质4的分子量相同。需要进一步做二级质谱研究。对应一级质谱图如9所示。rrt1.96色谱峰,测得的[m-h]-为483.3,分子量为484.3。对照usp34中的杂质为假单胞酸c,化学式为c26h44o8,化学名称为:9-((e)-4-((2s,3r,4r,5s)-3,4-dihydroxy-5-((4r,5s,e)-5-hydroxy-4-methylhex-2-enyl)tetrahydro-2h-pyran-2-yl)-3-methylbut-2-enoyloxy)nonanoicacid。对应一级质谱图如10所示,结构式如式iv所示。rrt2.13色谱峰,测得的[m-h]-为527.3,分子量为528.3。对照usp34中的杂质为假单胞酸e,化学式为c28h48o9,化学名称为:11-((e)-4-((2s,3r,4r,5s)-3,4-dihydroxy-5-(((2s,3s)-3-hydroxybutan-2-yl)oxiran-2-yl)methyl)tetrahydro-2h-pyran-2-yl)-3-methylbut-2-enoyloxy)undecanoicacid。对应一级质谱图11所示,结构式如式v所示。二级质谱分析二级质谱图如图12所示。rrt0.62色谱峰,在二级质谱获得[m-h]-173.2和325.2的碎片,见图13。其裂解过程见式vi所示。根据其二级质谱碎片和裂解过程分析,对照usp34中的杂质为莫匹罗星杂质1,化学式为c26h44o9,化学名称为:9-((e)-4-((2r,3as,6s,7s,8ars)-2-((1rs,2s,3s)-1,3-dihydroxy-2-methylbutyl)-7-hydroxyhexahydro-2h-furo(3,2-c)pyran-6-yl)-3-methylbutrrt0.65色谱峰,在二级质谱获得[m-h]-173.2、229.2和325.2的碎片,见图14。其裂解过程见式vii所示。根据其二级质谱碎片和裂解过程分析,对照usp34中的杂质为莫匹罗星杂质2,化学式为c26h44o9,化学名称为:9-((e)-4-((2r,3rs,4as,7s,8s,8ar)-3,8-dihydroxy-2-((2s,3s)-3-hydroxybutan-2-yl)octahydropyrano(3,2-c)pyran-7-yl)-3-methylbut-2-enoyloxy)nonanoicacid。rrt1.12和rrt1.24的色谱峰,在二级质谱获得[m-h]-173.2的碎片,见图15、图16。其裂解过程见式viii示。参照usp34莫匹罗星乳膏质量标准,用已知杂质的相对保留时间对杂质进行定性定位,对应的杂质见下表1:表1杂质定位表名称相对保留时间假单胞酸f0.36莫匹罗星杂质10.6莫匹罗星杂质20.63假单胞酸d0.75假单胞酸b0.9莫匹罗星1.0莫匹罗星杂质31.15莫匹罗星杂质41.23假单胞酸c2.03假单胞酸e2.24实施例二色谱测试条件确定过程。莫匹罗星对照品:中国药品生物制品检定所,批号130568-200501。试验样品:新型莫匹罗星软膏,湖北成田制药股份有限公司研制。规格:5g∶含0.1g莫匹罗星。2.1检测波长的确定usp34版莫匹罗星乳膏质量标准中控制了9个已知杂质,采用杂质的相对保留时间对杂质进行定性定位,由于无法获得杂质对照品,采用二极管阵列检测器,在200~400nm波长内扫描,提取每个杂质峰紫外吸收光谱图,采用莫匹罗星对照品溶液,获取莫匹罗星的紫外吸收图,从而确定检测波长。2.1.1仪器agilent1200高效液相色谱系统;agilent1200chemstation工作站;g1315cdad检测器;g1367d自动进样器2.1.2色谱条件色谱条件如下:色谱柱:zorbaxc8(250mm×4.6mm,5μm)检测波长:240nm流速:1ml/min进样体积:20μl柱温:35℃流动相:流动相c:0.1m醋酸铵∶四氢呋喃=75∶25流动相d:0.1m醋酸铵∶四氢呋喃=70∶30时间(min)流动相c(%)流动相d(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度2.1.3供试品溶液莫匹罗星软膏样品(批号:090501)置100℃环境中放置48小时,取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为供试品溶液(每1ml含约莫匹罗星5mg)。2.1.4对照品溶液取莫匹罗星对照品10mg,精密称定,置10ml量瓶中,加四氢呋喃溶液5ml,超声溶解,加醋酸钠溶液至刻度,摇匀;精密量取上述溶液1ml,置10ml量瓶中,用醋酸钠和四氢呋喃混合溶液稀释至刻度,摇匀,作为对照品溶液(每1ml含约莫匹罗星100μg)。2.1.5测试分别量取供试品溶液、对照品溶液各20μl,注入液相色谱仪,在200~400nm波长内扫描,记录色谱图,提取各杂质峰及主峰的紫外吸收光谱图。莫匹罗星及已知杂质紫外吸收光谱图如图17至图23所示。以上紫外吸收光谱图结果显示,莫匹罗星及各已知杂质均在波长221nm处有最大吸收,靠近紫外末端,而色谱试剂四氢呋喃越靠近紫外末端,其吸收越大,易引起色谱基线漂移。比较220nm、230nm、240nm波长处色谱图,杂质个数相同。为了减小基线漂移的影响,参照usp34莫匹罗星乳膏质量标准,选择波长240nm作为有关物质检测波长。2.2系统适用性试验2.2.1仪器依利特1201液相色谱仪2.2.2色谱条件色谱条件:见2.1.2。2.2.3溶液制备2.2.3.1供试品溶液取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为供试品溶液(每1ml含约莫匹罗星5mg)。2.2.3.2对照溶液精密量取供试品溶液1ml,置50ml量瓶中,用醋酸钠和四氢呋喃混合溶液稀释至刻度,摇匀,作为对照溶液(每1ml含约莫匹罗星100μg)。2.2.3.3空白辅料溶液取空白基质约2.5g,按供试品溶液制备方法制备空白辅料溶液。2.3.4系统适用性试验精密量取对照溶液20μl注入液相色谱仪,调节仪器灵敏度,使对照溶液的主峰峰高为满量程的20%。莫匹罗星的柱效应不小于7000,莫匹罗星峰的拖尾因子应不大于1.75,重复进样5次,莫匹罗星的相对标准偏差应小于2%。在上述色谱条件下,精密量取供试品溶液20μl注入液相色谱仪,假单胞酸d与莫匹罗星的分离度不得小于3。表2。表2系统适应性试验结果结论:此条件下莫匹罗星的保留时间约为20min,对照溶液色谱图中,理论板数按莫匹罗星计为50488,莫匹罗星峰的拖尾因子为1.16,重复进样5次,莫匹罗星峰面积相对标准偏差为0.94%,供试品溶液色谱图中,假单胞酸d与莫匹罗星的分离度为4.12,各杂质间分离度大于1.0,符合系统适应性要求。2.3.5空白辅料干扰试验精密量取空白辅料溶液20μl,注入液相色谱仪,记录色谱图,考察空白辅料对测定的干扰情况。在4min以前空白辅料有一吸收峰,与溶剂峰靠近,由于溶剂峰、基质峰位置靠前,且峰形尖锐,与莫匹罗星主峰及杂质峰均分离较好,对杂质测定无干扰。2.3.6专属性试验对新型莫匹罗星软膏进行强酸、强碱、氧化、强光、高温破坏性试验,考察拟定的色谱条件能否有效分离各降解产物。2.3.6.1破坏性样品溶液制备①酸破坏样品溶液:取供试品溶液10ml,加1mol/l盐酸1ml,放置1天,加1mol/l氢氧化钠1ml,摇匀,滤过。同时取空白辅料溶液10ml进行空白试验。②碱破坏样品溶液:取供试品溶液10ml,加1mol/l氢氧化钠1ml,放置1天,加1mol/l盐酸1ml,摇匀,滤过。同时取空白辅料溶液10ml进行空白试验。③氧化破坏样品溶液:取供试品溶液10ml,加30%双氧水1ml,放置2天,摇匀,滤过。同时取空白辅料溶液10ml进行空白试验。④光照破坏样品溶液:取供试品溶液10ml,在4500lx光照条件下,放置2天,摇匀,滤过。同时取空白辅料溶液10ml进行空白试验。⑤高温破坏样品溶液:取供试品溶液10ml,在105℃条件下,放置1天,摇匀,滤过。同时取空白辅料溶液10ml进行空白试验。2.3.6.2试验结果取上述破坏样品溶液各20μl,分别注入液相色谱仪,记录色谱图,测试结果见表3~7。表3酸破坏性试验结果表4碱破坏性试验结果组份名称保留时间(min)相对保留时间分离度碱破坏杂质13.880000.17----碱破坏杂质24.721670.214.34碱破坏杂质34.977500.221.15碱破坏杂质4(假单胞酸f)7.610000.349.30碱破坏杂质59.600000.434.98碱破坏杂质610.167500.461.28碱破坏杂质712.900830.586.96碱破坏杂质8(莫匹罗星杂质1)13.363330.600.91碱破坏杂质9(莫匹罗星杂质2)14.294170.641.40碱破坏杂质10(假单胞酸d)16.777500.763.74碱破坏杂质1121.089170.956.59莫匹罗星主峰22.195001.001.25碱破坏杂质12(莫匹罗星杂质3)25.652501.163.55碱破坏杂质13(莫匹罗星杂质4)27.273331.232.00碱破坏杂质14(假单胞酸c)42.205831.9017.07表5高温破坏性试验结果组份名称保留时间(min)相对保留时间分离度高温破坏杂质14.709170.22---高温破坏杂质24.959170.231.19高温破坏杂质3(假单胞酸f)7.566670.359.12高温破坏杂质49.531670.445.08高温破坏杂质510.089170.471.28高温破坏杂质611.505000.543.36高温破坏杂质712.083330.561.47高温破坏杂质812.627500.591.33高温破坏杂质9(莫匹罗星杂质1)13.184170.611.06高温破坏杂质10(莫匹罗星杂质2)13.972500.651.16高温破坏杂质1115.478330.722.44高温破坏杂质12(假单胞酸d)16.555830.771.86高温破坏杂质1318.270830.852.96高温破坏杂质1418.923330.881.23高温破坏杂质15(假单胞酸b)19.744170.921.26高温破坏杂质1620.545830.961.25莫匹罗星主峰21.503331.001.00高温破坏杂质17(莫匹罗星杂质3)23.949171.112.34高温破坏杂质1825.345831.181.85高温破坏杂质19(莫匹罗星杂质4)26.848331.251.83高温破坏杂质20(假单胞酸c)41.936671.9516.43高温破坏杂质21(假单胞酸e)47.565002.214.32表6氧化破坏性试验结果组份名称保留时间(min)相对保留时间分离度氧化破坏杂质1(假单胞酸f)7.245000.35----氧化破坏杂质29.110830.455.15氧化破坏杂质39.625830.471.29氧化破坏杂质4(莫匹罗星杂质1)13.024170.647.21氧化破坏杂质5(莫匹罗星杂质2)13.703330.671.24氧化破坏杂质614.930000.732.14氧化破坏杂质7(假单胞酸d)15.828330.771.48氧化破坏杂质817.601670.863.07氧化破坏杂质918.268330.891.31氧化破坏杂质10(假单胞酸b)18.960000.931.30氧化破坏杂质1119.649170.961.34莫匹罗星主峰20.425001.000.80氧化破坏杂质12(莫匹罗星杂质3)23.912921.173.40氧化破坏杂质13(莫匹罗星杂质4)25.400831.242.69氧化破坏杂质1425.925001.271.03氧化破坏杂质15(假单胞酸c)40.476671.9819.37氧化破坏杂质16(假单胞酸e)46.014172.255.08表7光照破坏性试验结果组份名称保留时间(min)相对保留时间分离度光照破坏杂质1(假单胞酸f)7.535830.36----光照破坏杂质29.478330.454.93光照破坏杂质310.054170.471.34光照破坏杂质4(莫匹罗星杂质1)13.549170.647.29光照破坏杂质5(莫匹罗星杂质2)14.245830.671.26光照破坏杂质615.522500.732.12光照破坏杂质7(假单胞酸d)16.466670.781.47光照破坏杂质817.495000.821.81光照破坏杂质918.169170.861.35光照破坏杂质1018.887500.891.35光照破坏杂质11(假单胞酸b)19.655830.931.43光照破坏杂质1220.428330.961.48莫匹罗星主峰21.227501.000.76光照破坏杂质13(莫匹罗星杂质4)25.402501.203.49光照破坏杂质1427.013331.272.18光照破坏杂质15(假单胞酸c)42.166671.9917.79光照破坏杂质16(假单胞酸e)47.812502.255.412.3.6.3试验结论根据以上测试结果,莫匹罗星在强酸、强碱、氧化、强光、高温条件下均有明显降解,在拟定的色谱条下,各降解产物均能与莫匹罗星主峰得到良好的分离。2.3.7检测限测定精密量取对照品溶液1ml,用醋酸钠和四氢呋喃混合溶液依次稀释至101、102、103等倍,精密量取各稀释级对照溶液20μl,依次注入液相色谱仪,记录色谱图,以信噪比为3∶1时的注入量为检测限。最小检出量为40ng。2.3.8耐用性试验改变色谱条件下某个参数,考察其耐用性。取供试品溶液20μl注入液相色谱仪,假单胞酸d与莫匹罗星的分离度不得小于3,各杂质峰之间的分离度不得小于1.0,与莫匹罗星主峰相邻的杂质峰与主峰之间的分离度不得小于0.75。2.3.8.1改变色谱柱型号①zorbaxeclipsexdb-c8(250mm×4.6mm,5μm)②zorbaxeclipseplusc8(250mm×4.6mm,5μm)③varianpursuit5c8(250mm×4.6mm,5μm)④cosmosil5c8-ms(250mm×4.6mm,5μm)⑤purospherstarrp-8e(250mm×4.6mm,5μm)试验结果见表8。表8色谱柱耐用性试验结果结论:用五根不同型号的色谱柱c8(250mm×4.6mm,5μm)进行耐用性考察,假单胞酸d与莫匹罗星的分离度分别为6.99、6.44、7.35、8.65、6.99,各杂质峰之间的分离度均大于1.0,与莫匹罗星主峰相邻的杂质峰与主峰之间的分离度均大于0.75,符合耐用性要求。9个已知杂质的相对保留时间与usp34基本相符,但未知杂质的相对保留时间相差较大,只有zorbaxeclipsexdb-c8、zorbaxeclipseplusc8的色图谱一致。前期关于已知杂质的定位研究中(液质联动法)采用的色谱柱是zorbaxeclipsexdb-c8(250mm×4.6mm,5μm),因此将色谱柱确定为zorbaxc8(250mm×4.6mm,5μm)。2.3.8.2改变流动相ph①ph5.7;②ph5.6;③ph5.8。表9。表9流动相ph耐用性试验结果结论:不推荐改变ph±0.1,因为降低ph导致莫匹罗星和假单胞酸c之间的杂质个数减少,ph升高导致假单胞酸d和莫匹罗星之间的杂质个数减少。缓冲液最佳ph是5.7。2.3.8.3改变流动相比例①流动相c:0.1m醋酸铵∶四氢呋喃=75∶25流动相d:0.1m醋酸铵∶四氢呋喃=70∶30②流动相c:0.1m醋酸铵∶四氢呋喃=77∶23流动相d:0.1m醋酸铵∶四氢呋喃=72∶28③流动相c:0.1m醋酸铵∶四氢呋喃=73∶27流动相d:0.1m醋酸铵∶四氢呋喃=68∶32④流动相c:0.1m醋酸铵∶四氢呋喃=76∶24流动相d:0.1m醋酸铵∶四氢呋喃=71∶29⑤流动相c:0.1m醋酸铵∶四氢呋喃=74∶26流动相d:0.1m醋酸铵∶四氢呋喃=69∶31试验结果见10表10流动相比例耐用性试验结果结论:改变流动相比例2%引起相对保留时间的重大改变,流动相比例增加2%导致假单胞酸d和莫匹罗星之间的杂质个数减少,流动相比例减小2%导致莫匹罗星和假单胞酸c之间的杂质个数减少。流动相组分只能改变1%。2.8.4改变柱温①柱温35℃;②柱温40℃③柱温30℃④柱温37℃⑤柱温33℃试验结果见表11表11柱温耐用性试验结果结论:不能改变柱温5℃,柱温升高5℃导致假单胞酸d和莫匹罗星之间的杂质个数减少。柱温降低5℃导致莫匹罗星和假单胞酸c之间的杂质个数减少,且主峰与前一杂质峰的分离度<0.75。可以改变柱温2℃。实施例三对新型莫匹罗星软膏产品三个批次的产品进行检测,分别为第一批次、第二批次及第三批次杂质含量检测。检测步骤如下:步骤a1:将莫匹罗星软膏样品制备成含量测试样品;莫匹罗星软膏样品置100℃环境中放置48小时,取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为定位测试样品(每1ml含约莫匹罗星5mg)。步骤a2:将所述含量测试样品进行高效色相色谱检测;色谱条件如下:色谱柱:zorbaxc8(250mm×4.6mm,5μm)检测波长:240nm流速:1ml/min进样体积:20μl柱温:35℃流动相:流动相c:0.1m醋酸铵∶四氢呋喃=75∶25流动相d:0.1m醋酸铵∶四氢呋喃=70∶30梯度表时间(min)流动相c(%)流动相d(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度步骤a3:将所述步骤a2的结果与杂质标准色谱图进行比对,得到所述莫匹罗星软膏杂质含量;其中,所述杂质标准色谱通过实施例一莫匹罗星软膏杂质定性定位方法检测得到,测试结果如表12所示。表12莫匹罗星软膏杂质测试结果实施例四对新型莫匹罗星软膏产品三个批次的产品进行检测,分别为第一批次、第二批次及第三批次杂质含量检测。检测步骤如下:步骤a1:将莫匹罗星软膏样品制备成含量测试样品;莫匹罗星软膏样品置100℃环境中放置48小时,取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为定位测试样品(每1ml含约莫匹罗星5mg)。步骤a2:将所述含量测试样品进行高效色相色谱检测;色谱条件如下:色谱柱:zorbaxc8(250mm×4.6mm,5μm)检测波长:240nm流速:1ml/min进样体积:20μl柱温:33℃流动相:流动相c:0.1m醋酸铵∶四氢呋喃=74∶26流动相d:0.1m醋酸铵∶四氢呋喃=69∶31梯度表时间(min)流动相c(%)流动相d(%)洗脱01000平衡0-61000单液等度6-35100→00→100线性梯度35-550100单液等度55-55.010→100100→0台阶梯度55.01-651000单液等度步骤a3:将所述步骤a2的结果与杂质标准色谱图进行比对,得到所述莫匹罗星软膏杂质含量;其中,所述杂质标准色谱通过实施例一莫匹罗星软膏杂质定性定位方法检测得到。实施例五对新型莫匹罗星软膏产品三个批次的产品进行检测,分别为第一批次、第二批次及第三批次杂质含量检测。检测步骤如下:步骤a1:将莫匹罗星软膏样品制备成含量测试样品;莫匹罗星软膏样品置100℃环境中放置48小时,取本品适量(约相当于莫匹罗星50mg),精密称定,置小烧杯瓶中,加四氢呋喃溶液5ml,搅拌溶解,加醋酸钠溶液5ml,摇匀,0.5μm微孔滤膜滤过,取续滤液作为定位测试样品(每1ml含约莫匹罗星5mg)。步骤a2:将所述含量测试样品进行高效色相色谱检测;色谱条件如下:色谱柱:zorbaxc8(250mm×4.6mm,5μm)检测波长:240nm流速:1ml/min进样体积:20μl柱温:37℃流动相:④流动相c:0.1m醋酸铵∶四氢呋喃=76∶24流动相d:0.1m醋酸铵∶四氢呋喃=71∶29梯度表步骤a3:将所述步骤a2的结果与杂质标准色谱图进行比对,得到所述莫匹罗星软膏杂质含量;其中,所述杂质标准色谱通过实施例一莫匹罗星软膏杂质定性定位方法检测得到。对比例一对实施例三同批次样品进行检测,检测步骤如下:莫匹罗星软膏(约相当于莫匹罗星50mg),加0.1mol/l磷酸二氢钠缓冲液(用1mol/l氢氧化钠溶液调节ph值至6.3)适量,制成每1ml含约莫匹罗星5mg的溶液。进行色谱检测,色谱条件按bp标准测试测试后比对:bp2009版莫罗星软膏控制了3个已知杂质,莫匹罗星杂质c(相对莫匹罗星保留时间约0.65)、莫匹罗星杂质d(相对莫匹罗星保留时间约0.5)、莫匹罗星杂质e(相对莫匹罗星保留时间约0.55),但在已知杂质定位液质联用研究中流速改变为1ml/min,引起已知杂质的相对保留时间与bp2009莫匹罗星软膏规定相差约0.1,因此将3个已知杂质的相对保留时间修订为:莫匹罗星杂质c(相对莫匹罗星保留时间约0.75)、莫匹罗星杂质d(相对莫匹罗星保留时间约0.6)、莫匹罗星杂质e(相对莫匹罗星保留时间约0.64),意味着按※标记的杂质按其他未知杂质控制,限度为1.5%。测试结果如表13所示。表13莫匹罗星软膏杂质测试结果本发明未采用杂质对照品进行对比,采用合理色谱质谱条件,便可控制9种杂质,后续便可合理的色谱测试直接进行比对即可,对莫匹罗星软膏的研制过程中杂质检测及控制提供了新的思路。对比例检出15个杂质,本发明方法检出16个杂质。以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1