利用高效液相色谱法测定驱虫药有效成分含量的方法与流程
本发明属于化学分析
技术领域:
,涉及一种利用高效液相色谱法测定驱虫药有效成分含量的方法。
背景技术:
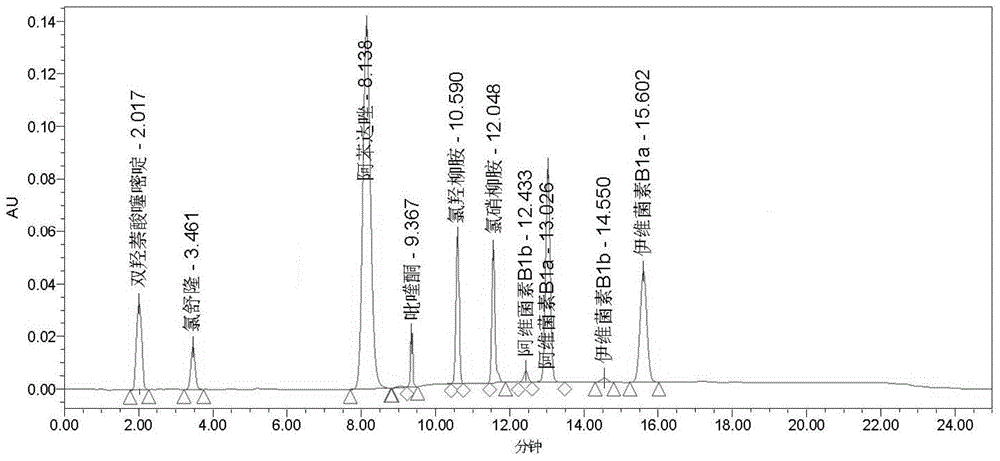
:动物寄生虫病是动物三大类疾病之一,寄生虫感染直接或间接影响动物的健康和发育。寄生虫根据种属分成不同的生物类群,不同种属对药物的敏感程度不一样,然而单个动物可同时感染一种或一种以上寄生虫感染,故不同的寄生虫感染选择不同的药物。目前市场的驱虫药多为复方制剂,含两种或两种以上的成分检测方法存在操作繁琐、耗时等缺点,虽然有少量文献报道同时测定驱虫药中某两种成分驱虫药的含量的方法,其仍然存在种类检测不全等缺点,而且迄今为止尚未见同时检测驱虫药中8种有效成分含量的方法,这8种有效成分包括阿苯达唑、阿维菌素、伊维菌素、氯舒隆、吡喹酮、双羟萘酸嘧啶、氯羟柳胺、氯硝柳胺。因此,研发一种利用高效液相色谱法同时测定驱虫药中8种有效成分含量的方法是本领域人员亟需解决的技术问题。技术实现要素:有鉴于此,本发明提供一种利用高效液相色谱法同时测定驱虫药中8种有效成分含量的方法,可广泛应用于医药与食品行业中复合驱虫产品的质量控制,有效地节约检测成本与时间。为了实现上述目的,本发明采用如下技术方案:一种利用高效液相色谱法测定驱虫药有效成分含量的方法,包括以下步骤:1)供试品溶液的制备取供试品,制成供试品溶液;2)对照品溶液的制备取阿苯达唑、阿维菌素、伊维菌素、氯舒隆、吡喹酮、双羟萘酸嘧啶、氯羟柳胺、氯硝柳胺精密称定,加甲醇溶解、稀释并定容;3)色谱条件色谱柱填充剂:十八烷基硅烷键合硅胶;流动相及洗脱方式:以缓冲溶液0.1mol/l醋酸钠溶液为流动相a,以甲醇为流动相b,以乙腈为流动相c,按以下程序进行梯度洗脱:0-6min:流动相为体积比60:40的流动相a-流动相c混合液;6-8min:流动相由体积比60:40的流动相a-流动相c混合液线性变化为体积比5:9:86的流动相a-流动相b-流动相c混合液;8-20min:流动相由体积比5:9:86的流动相a-流动相b-流动相c混合液;20-22min:流动相由体积比5:9:86的流动相a-流动相b-流动相c混合液线性变为体积比60:40的流动相a-流动相c混合液;22-25min:流动相为体积比60:40的流动相a-流动相c混合液;检测波长:按以下程序采用变化波长:0-6min:检测波长为210nm;6-10min:检测波长为295nm;10-15min:检测波长为210nm;15-25min:检测波长为243nm;柱温:35℃;流速:1ml/min;进样量:20ul;4)测定精密量取供试品溶液注入液相色谱仪,记录色谱图;另精密量取对照品溶液注入液相色谱仪,同法测定;按外标法以峰面积分别计算供试品中各有效成分的含量。进一步,步骤4)所述各有效成分的含量计算公式如下式所示:式中:x:以标示量表示的相对百分含量(%);ar:对照品的峰面积;mr:对照品的称取量(mg);ax:供试品的峰面积;mx:供试品的取量(mg);c:干燥对照品含量(%)。进一步,步骤1)上述供试品溶液的制备方法包括以下步骤:供试品为固体样品:取供试品,研细,精密称定,置100ml棕色量瓶中,加入甲醇超声15min,加入甲醇稀释至刻度,摇匀,滤过,所得滤液作为供试品溶液;或,供试品为非水溶剂注射液:精密量取供试品,置100ml棕色量瓶中,用甲醇稀释至刻度,摇匀,作为供试品溶液;或,供试品为除含水注射液外的其它液体样品:精密量取供试品,置100ml棕色量瓶中,加入甲醇溶解稀释至刻度,摇匀,滤过,所得滤液作为供试品溶液。进一步,步骤2)上述对照品溶液的制备方法包括以下步骤:精密称取阿苯达唑25mg、阿维菌素10mg、伊维菌素10mg、氯舒隆10mg、吡喹酮10mg、双羟萘酸嘧啶50mg、氯羟柳胺25mg、氯硝柳胺25mg,置100ml棕色量瓶中,加甲醇10ml超声使溶解,用甲醇稀释至刻度,摇匀,作为对照品溶液。本发明的有益效果在于:本发明采用反相高效液相色谱法同时测定驱虫药中8种有效成分的含量,采用价廉易得的十八烷基硅烷键合硅胶为填充剂,以常用缓冲溶液-甲醇-乙腈为流动相进行梯度洗脱,联合流动相的梯度流速和紫外检测器的程序变化波长,在25分钟内可实现驱虫药中8种有效成分的完好分离与定量分析,方法专属性强、精密度高、稳定性好、准确度高,操作简便,试剂与耗材价廉易得,适用于医药与农化行业中复合驱虫产品或杀虫剂的质量控制,可有效地节约检测成本与时间。附图说明图1为含8种有效成分对照品溶液的高效液相色谱图。图2为市售复方驱虫片的高效液相色谱图。图3为市售复方驱虫口服液的高效液相色谱图。具体实施方式结合附图和以下实施例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。一种利用高效液相色谱法测定驱虫药有效成分含量的方法,包括以下步骤:1)供试品溶液的制备取供试品,制成供试品溶液;2)对照品溶液的制备取阿苯达唑、阿维菌素、伊维菌素、氯舒隆、吡喹酮、双羟萘酸嘧啶、氯羟柳胺、氯硝柳胺精密称定,加甲醇溶解、稀释并定容;3)色谱条件色谱柱填充剂:十八烷基硅烷键合硅胶;流动相及洗脱方式:以0.1mol/l醋酸钠溶液为流动相a,以甲醇为流动相b,以乙腈为流动相c,按以下程序进行梯度洗脱:0-6min:流动相为体积比60:40的流动相a-流动相c混合液;6-8min:流动相由体积比60:40的流动相a-流动相c混合液线性变化为体积比5:9:86的流动相a-流动相b-流动相c混合液;8-20min:流动相由体积比5:9:86的流动相a-流动相b-流动相c混合液;20-22min:流动相由体积比5:9:86的流动相a-流动相b-流动相c混合液线性变为体积比60:40的流动相a-流动相c混合液;22-25min:流动相为体积比60:40的流动相a-流动相c混合液;检测波长:按以下程序采用变化波长:0-6min:检测波长为210nm;6-10min:检测波长为295nm;10-15min:检测波长为210nm;15-25min:检测波长为243nm;柱温:35℃;流速:1ml/min;进样量:20ul;4)测定精密量取供试品溶液注入液相色谱仪,记录色谱图;另精密量取对照品溶液注入液相色谱仪,同法测定;按外标法以峰面积分别计算供试品中各有效成分的含量。步骤4)所述各有效成分的含量计算公式如下式所示:式中:x:以标示量表示的相对百分含量(%);ar:对照品的峰面积;mr:对照品的称取量(mg);ax:供试品的峰面积;mx:供试品的取量(mg);c:干燥对照品含量(%)。步骤1)供试品溶液的制备方法包括以下步骤:供试品为固体样品:取供试品,研细,精密称定,置100ml棕色量瓶中,加入甲醇超声15min,加入甲醇稀释至刻度,摇匀,滤过,所得滤液作为供试品溶液;或,供试品为非水溶剂注射液:精密量取供试品,置100ml棕色量瓶中,用甲醇稀释至刻度,摇匀,作为供试品溶液;或,供试品为除含水注射液外的其它液体样品:精密量取供试品,置100ml棕色量瓶中,加入甲醇溶解稀释至刻度,摇匀,滤过,所得滤液作为供试品溶液。步骤2)对照品溶液的制备方法包括以下步骤:精密称取阿苯达唑25mg、阿维菌素10mg、伊维菌素10mg、氯舒隆10mg、吡喹酮10mg、双羟萘酸嘧啶50mg、氯羟柳胺25mg、氯硝柳胺25mg,置100ml棕色量瓶中,加甲醇10ml超声使溶解,用甲醇稀释至刻度,摇匀,作为对照品溶液。含8种有效成分对照品溶液的高效液相色谱图见图1。实施例1以市售复合驱虫片作为供试品,其标示每片含有吡喹酮80mg、阿苯达唑90mg、双羟萘酸噻嘧啶50mg;利用高效液相色谱法测定驱虫药有效成分含量的方法,包括以下步骤:1)供试品溶液的制备:取供试品适量,研细,精密称定,置100ml棕色量瓶中,加10ml超声10分钟稀释至刻度,摇匀,滤过,所得滤液作为供试品溶液。步骤2)-4)与实施例1相同,结果见图2。由图2可知,该复合驱虫片所含有吡喹酮80mg、阿苯达唑90mg、双羟萘酸嘧啶50mg,其标示百分含量分别为:吡喹酮99.4%、阿苯达唑98.2%、双羟萘酸噻嘧啶96.4%。实施例2以市售复合驱虫口服液作为供试品,其标示每ml含有阿苯达唑100mg、氯羟柳胺35mg、吡喹酮50mg、双羟萘酸噻嘧啶25mg;利用高效液相色谱法测定驱虫药有效成分含量的方法,包括以下步骤:1)供试品溶液的制备:精密量取供试品5ml,置100ml量瓶中,加甲醇10ml超声10分钟,用甲醇稀释至刻度,摇匀,作为供试品溶液。步骤2)-4)与实施例1相同,结果见图3。由图3可知,该复合驱虫口服液含有阿苯达唑,氯羟柳胺,吡喹酮、双羟萘酸噻嘧啶,其标示百分含量分别为:阿苯达唑97.2%,氯羟柳胺99.4%,吡喹酮96.4%、双羟萘酸噻嘧啶98.4%。含量测定方法的验证1.专属性考察驱虫药8种有效成分为阿苯达唑、阿维菌素、伊维菌素、氯舒隆、吡喹酮、双羟萘酸嘧啶、氯羟柳、氯硝柳胺,分别精密量取对照品溶液与驱虫药8种有效成分的各阴性供试液各20μl,注入液相色谱仪,记录色谱图,对照品溶液的色谱图见图1。通过比较对照品溶液和驱虫药8种有效成分的各阴性供试液的色谱图,确认8种有效成分各自的色谱峰平均保留时间见表1。比较对照品溶液和8种有效成分的各阴性供试液的色谱图还可以看出,各成分色谱峰的保留时间一致,说明8种有效成分色谱峰在保留时间位置上阴性供试液没有明显干扰,进一步说明在本发明所述色谱条件下,8种有效成分之间可以实现完好分离。表18种有效成分的色谱峰平均保留时间2.线性关系考察精密量取对照品溶液5μl、10μl、15μl、30μl、50μl分别注入液相色谱仪,记录色谱图。以进样量x(mg)为横坐标,峰面积y为纵坐标作图绘制标准曲线,计算8种有效成分的线性回归方程与相关系数(r),结果见表2。由表2可知,8种有效成分在线性范围内,峰面积与浓度均呈现良好的线性关系。表2线性关系考察结果3.精密度考察精密量取对照品溶液20μl注入液相色谱仪,记录色谱图,重复6次,以各成分色谱峰的6次测得峰面积计算相对标准偏差(rsd),结果见表3。由表3可知,8种有效成分色谱峰的峰面积rsd均符合精密度要求,说明本发明方法精密度良好。表3精密度考察结果有效成分rsd(%)氯舒隆2.12双羟萘酸噻嘧啶1.22阿苯达唑1.14吡喹酮1.50氯羟柳胺1.45氯硝柳胺1.06阿维菌素1.17伊维菌素1.214.稳定性考察制备对照品溶液,分别于0、1、2、4、6、8、12小时精密量取对照品溶液20μl注入液相色谱仪,记录色谱图。以各有效成分色谱峰的7次测得峰面积计算rsd。结果见表4。由表4可知,8种有效成分色谱峰的峰面积rsd均符合精密度要求,说明本发明方法稳定性良好。表4稳定性考察结果有效成分rsd(%)氯舒隆2.12双羟萘酸噻嘧啶1.10阿苯达唑1.20吡喹酮1.74氯羟柳胺1.45氯硝柳胺1.87阿维菌素1.09伊维菌素1.015.准确度考察加样回收率试验:精密量取已知各成分含量的供试品溶液9份,每3份为1组,每组均按照低、中、高三个浓度分别精密加入对照品溶液4ml、5ml、6ml,摇匀,精密量取20μl分别注入液相色谱仪,记录色谱图,计算8种有效成分的平均加样回收率和rsd,结果见表5。由表5可知,8种有效成分的平均加样回收率均大于97.5%,rsd不大于2.2%,说明本发明方法准确度高。表5准确度考察结果以上内容是结合具体的优先实施方式对本发明所作的进一步说明,不能认定本发明的具体实施只局限于这些说明,对于发明所属
技术领域:
的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演和替换,都应当视为属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1