复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法与流程
本发明属于药物化学检测技术领域,具体涉及到一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法。
背景技术:
丙氨酰谷氨酰胺是肠外营养产品中非常重要的一种二肽,可在人体内快速分解为人体必须的两种氨基酸:丙氨酸和谷氨酸,且无毒副作用,其在肠外营养中的应用正在日益受到重视。丙氨酰谷氨酰胺在高温等条件的作用下会降解,对于复方产品来说,使用常规的hplc(高效液相色谱)法进行检测时,复杂的氨基酸组分对于丙氨酰谷氨酰胺四种杂质l-焦谷氨酸、环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸)的检测会产生干扰。simone[1]等人使用lc-esi-ms/ms对肠外氨基酸注射液中的丙氨酰谷氨酰胺杂质进行检测,但是lc-esi-ms/ms设备昂贵,一般的实验室不具备该条件。simone[2]等人使用二维反向色谱×hilic分离模式,it-ms与cad做检测器分析肠外氨基酸注射液中的丙氨酰谷氨酰胺杂质,该法设备昂贵,操作复杂,一般实验室不具备该条件。
非专利文献:[1]simoneschiesel,michaellammerhofer,alexanderleitner,wolfganglindner.journalofchromatographya,1259(2012)111-120.
[2]simoneschiesel,michaellammerhofer,wolfganglindner.journalofchromatographya,1259(2012)100-110.
技术实现要素:
本发明所要解决的技术问题在于针对上述现有技术的不足,提供了一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法。本发明的检测方法简便、快速、成本低廉,使用阳离子交换树脂吸附去除复方氨基酸注射液中的氨基酸组分,方便后续丙氨酰谷氨酰胺杂质l-焦谷氨酸、环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸)的测定,而不用担心由氨基酸组分造成的干扰,测定过程中稀释剂与流动相均为水相,不涉及任何有机溶剂的使用,环境友好且节约成本。
为解决上述技术问题,本发明采用的技术方案是:一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)配制l-焦谷氨酸对照品溶液;
(2)将适量复方氨基酸注射液注入填充了阳离子交换树脂的层析柱内,以超纯水为洗脱溶剂进行淋洗,收集洗脱液,即得供试品溶液;
(3)确定高效液相色谱条件:
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,规格为50~250mm×2.1~4.6mm,填料粒径为1.7~5μm;
流动相:5~50mmol/l,磷酸氢二铵,ph1.9~2.1;
流速:0.9~1.1ml/min;
柱温:23~27℃;
检测波长:200~220nm;
进样体积:5~50μl;
(4)采用高效液相色谱法对l-焦谷氨酸对照品溶液及复方氨基酸注射液供试品溶液进行检测,记录相应色谱图;
(5)根据实验计算出环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸)三种杂质对l-焦谷氨酸的相对保留时间及响应的校正因子,再根据l-焦谷氨酸对照品的峰面积计算供试品溶液中丙氨酰谷氨酰胺杂质含量。
进一步地,步骤(1)中配制对照品溶液时采用的溶剂为纯水。
进一步地,步骤(2)中复方氨基酸注射液采用阳离子交换树脂的洗脱时保证氨基酸组分全部被阳离子树脂吸附去除,l-焦谷氨酸、环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸)四种杂质全部被洗脱出来。
进一步地,步骤(3)中所述色谱柱的规格为4.6mm×250mm,粒径为5μm。
进一步地,步骤(3)中所述流动相为浓度为10mmol/l的(nh4)2hpo4,ph值为2.0。
进一步地,步骤(3)中所述流速设置为1ml/min。
进一步地,步骤(3)中所述柱温设置为25℃。
进一步地,步骤(3)中所述检测波长设置为205nm。
进一步地,步骤(3)中所述进样量设置为25μl。
与现有技术相比,本发明的效果和益处如下:
(1)本发明的检测方法首先使用阳离子交换树脂吸附去除复方氨基酸注射液中的氨基酸组分,然后使用常规反相hplc测定丙氨酰谷氨酰胺杂质,使用阳离子交换树脂吸附去除复方氨基酸注射液中的氨基酸组分方便后续准确、快速的使用常规hplc测定丙氨酰谷氨酰胺杂质l-焦谷氨酸、环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸),而不用担心氨基酸组分造成的干扰。
(2)选取价格低廉且易于获得的l-焦谷氨酸作为标准物质,通过实验计算出环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸和环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间及响应的校正因子,使得在日常检测中仅需配制l-焦谷氨酸对照品溶液,便可准确检测出样品中四种杂质的含量,大大节约时间与成本。
(3)实验中稀释剂与流动相均为水相,不涉及任何有机溶剂的使用,环境友好且节约成本。
附图说明
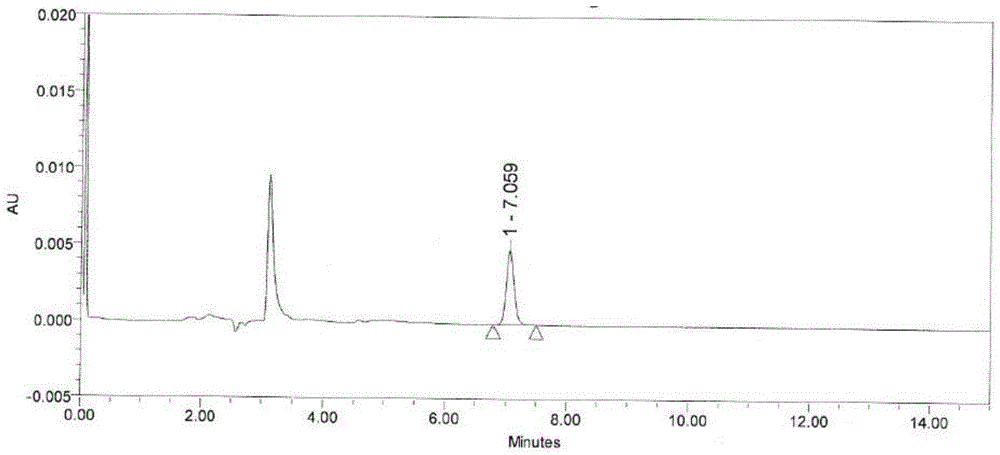
图1是本发明l-焦谷氨酸对照品溶液的hplc色谱图。
图2是本发明实施例1复方氨基酸注射液供试品溶液的hplc色谱图。
图3是本发明实施例2复方氨基酸注射液供试品溶液的hplc色谱图。
图4是本发明实施例3复方氨基酸注射液供试品溶液的hplc色谱图。
图5是本发明实施例4复方氨基酸注射液供试品溶液的hplc色谱图。
图6是本发明实施例5复方氨基酸注射液供试品溶液的hplc色谱图。
图7是本发明实施例6复方氨基酸注射液供试品溶液的hplc色谱图。
图8是本发明实施例7复方氨基酸注射液供试品溶液的hplc色谱图。
附图标记说明:1:l-焦谷氨酸;2:环-(l-丙氨酰-谷氨酰胺);3:l-焦谷氨酰-l-丙氨酸;4:环-(l-丙氨酰-谷氨酸)。
具体实施方式
下面结合具体实施例对本发明的技术方案作进一步说明。
实施例1
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.0;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.0ml/min;
柱温:25℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图1-2所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例2
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.0;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.0ml/min;
柱温:23℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图3所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例3
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.0;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.0ml/min;
柱温:27℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图4所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别约为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例4
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.0;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:0.9ml/min;
柱温:25℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图5所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例5
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液。
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.0;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.1ml/min;
柱温:25℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图6所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例6
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为1.9;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.0ml/min;
柱温:25℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图7所示;
步骤四、根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
实施例7
一种复方氨基酸注射液中丙氨酰谷氨酰胺杂质的检测方法,具体包括以下步骤:
(1)精密称取l-焦谷氨酸对照品56mg置于100ml量瓶内,用适量水溶解并稀释至刻度,得对照品贮备液,精密移取l-焦谷氨酸对照品贮备液1ml于250ml容量瓶内,以水稀释至刻度,摇匀,即得对照品工作溶液;
(2)称3.5gdowex50wx8树脂至50ml烧杯内,用水浸泡1h,然后小心填充至玻璃层析柱内,用30ml水进行淋洗,精密吸取2ml样品至层析柱内,用水淋洗,收集洗脱液至100ml容量瓶内,即得供试品溶液;
(3)确定高效液相色谱条件:
流动相:10mmol/l的(nh4)2hpo4,ph值为2.1;
色谱柱:亲水改性的烷基硅烷化学键合硅胶柱,粒径5μm,规格4.6×250mm;
检测波长:205nm;
流速:1.0ml/min;
柱温:25℃;
进样体积:25μl;
(4)采用hplc对l-焦谷氨酸对照品工作溶液及复方氨基酸供试品溶液进行分析并记录相应色谱图,如图8所示;
(5)根据对照品及供试品中杂质峰面积计算各杂质含量,计算公式如下:
式中
cs:样品中待测杂质含量(mg/ml);
cst:对照品工作溶液中l-焦谷氨酸的含量(mg/ml);
as:供试品中待测杂质峰面积;
ast:对照品工作溶液中l-焦谷氨酸色谱峰的面积均值;
d:样品稀释倍数,50;
f:相对于l-焦谷氨酸的校正因子。其中,环-(l-丙氨酰-谷氨酰胺)、l-焦谷氨酰-l-丙氨酸、环-(l-丙氨酰-谷氨酸)相对于l-焦谷氨酸的相对保留时间分别为:1.29、1.44、1.92;相对校正因子分别为:0.39、0.60、0.39。
以上仅就本发明较佳的实施例作了说明,但不能理解为是对权利要求的限制。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求所定义的范围。
- 还没有人留言评论。精彩留言会获得点赞!