一种同时测定格列吡嗪类药物中8种化合物含量的检测方法与流程
本发明属于检测
技术领域:
,具体涉及一种同时测定格列吡嗪类药物中8种化合物含量的检测方法。该方法可以用于测定格列吡嗪合成中间体、格列吡嗪原料药、含格列吡嗪药物中8种化合物的同时测定。
背景技术:
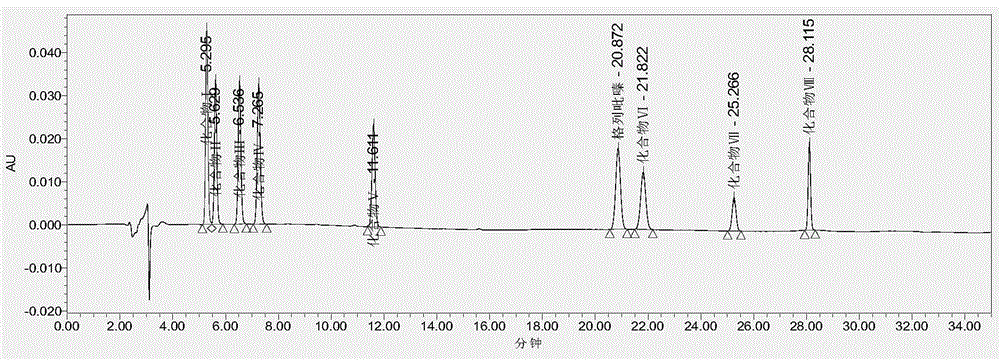
:格列吡嗪是一种在临床上广泛使用的降血糖药物。该药物属于化学合成药物,不可避免存在其他杂质,这些杂质主要来源于三个方面。一方面是合成过程中发生的副反应生成的化合物;另一方面是合成的起始化合物、中间产物残留;再一方面是原料药或者药物制剂在储存和运输过程产生的杂质。4-[2-(5-甲基吡嗪-2-甲酰氨基)乙基]苯磺酰胺(化合物ⅰ)、6-甲基-n-[2-(4-氨磺酰基苯基)乙基]吡嗪-2-羧酰胺(化合物ⅱ)、[2-(4-氨磺酰苯基)乙基]氨基甲酸乙酯(化合物ⅲ)、[4-[2-(5-甲基吡嗪-2-甲酰胺基)乙基]苯-1-磺酰基]氨基甲酸甲酯(化合物ⅳ)、4-[2-[(环己基氨甲酰)氨基]乙基]苯-1-磺胺、n-[2-[4-[(环己基氨甲酰)氨磺酰]苯基]乙基]-6-甲基吡嗪-2-甲酰胺(化合物ⅴ)、乙基[2-[4-[(环己基氨基甲酰基)氨磺酰基]苯基]乙基]氨基甲酸酯(化合物ⅶ)、n-(环己基氨甲酰)-4-[2-[(环己基氨甲酰)氨基]乙基]苯-1-磺胺(化合物ⅷ)为格列吡嗪合成过程引入或储存过程中降解的8种杂质化合物。化合物ⅰ:名称和分子式c14h16n4o3s320.374-[2-(5-甲基吡嗪-2-甲酰氨基)乙基]苯磺酰胺化合物ⅱ:名称和分子式c14h16n4o3s320.376-甲基-n-[2-(4-氨磺酰基苯基)乙基]吡嗪-2-羧酰胺化合物ⅲ:名称和分子式c11h16n2o4s272.32[2-(4-氨磺酰苯基)乙基]氨基甲酸乙酯化合物ⅳ:名称和分子式c16h18n4o5s378.40[4-[2-(5-甲基吡嗪-2-甲酰胺基)乙基]苯-1-磺酰基]氨基甲酸甲酯化合物ⅴ:名称和分子式c15h23n3o3s325.434-[2-[(环己基氨甲酰)氨基]乙基]苯-1-磺胺化合物ⅵ:名称和分子式c21h27n5o4s445.54n-[2-[4-[(环己基氨甲酰)氨磺酰]苯基]乙基]-6-甲基吡嗪-2-甲酰胺化合物ⅶ:名称和分子式c18h27n3o5s397.49乙基[2-[4-[(环己基氨基甲酰基)氨磺酰基]苯基]乙基]氨基甲酸酯化合物ⅷ:名称和分子式c22h34n4o4s450.60n-(环己基氨甲酰)-4-[2-[(环己基氨甲酰)氨基]乙基]苯-1-磺胺通过测定8种杂质化合物的含量,可以对格列吡嗪原料的合成进行有效的过程监控,也可以对原料药和含格列吡嗪药物的进行有效质量控制和评价。目前尚未见同时测定该8种化合物的详细定量测定方法报道。《中国药典》、《欧洲药典》、《美国药典》等,均收载了格列吡嗪有关物质测定方法,其中《欧洲药典》收载了格列吡嗪中可能存在的8种杂质,但《中国药典》、《欧洲药典》、《美国药典》均采用了主成分自身对照法,该方法是一种限度检查方式,由于未使用杂质对照品溶液,所以均不能对8种杂质进行准确定量。专利cn111679027a公开了一种通过高效液相色谱法分离测定格列吡嗪及其杂质的方法和用途,所述测定方法采用十八烷基硅烷键合硅胶为填充剂的色谱柱,其检测条件如下:色谱条件:色谱柱为十八烷基硅烷键合硅胶色谱柱,流动相为:0.010~0.018mol/l磷酸二氢钾-乙腈-甲醇,用磷酸调节ph值至3.5~4.0,柱温:20~30℃,进样量:15~30μl,进样浓度:0.0003~2mg/ml,流速:0.8~1.2ml/min,检测波长:225nm,梯度洗脱。洗脱梯度为阶段va相(%)vb相(%)vc相(%)洗脱时间06525107min15030205min24035256min33045256min溶液配制:稀释剂:0.015mol/l磷酸二氢钾(用磷酸调节ph值至3.7)-乙腈-甲醇(65∶25∶10)(即初始配比流动相)格列吡嗪与各杂质对照品储备溶液(杂质a~l):取格列吡嗪和杂质对照品各适量,分别置不同的50ml量瓶中,加稀释剂溶解并稀释至50ml,摇匀,定量稀释制成每1ml中含格列吡嗪和各杂质100μg的溶液。取格列吡嗪与各杂质对照品储备溶液(杂质a~l)作定位溶液。分离度溶液:取格列吡嗪对照品约20mg,精密称定,置50ml量瓶中,加甲醇20ml溶解,精密量取各杂质对照品储备溶液适量,置同一50ml量瓶中,用稀释剂定量稀释制成每1ml含格列吡嗪0.4mg、杂质a1.2μg、杂质e0.4μg、其余各杂质0.6μg的混合溶液,作为分离度溶液。测定:精密量取定位溶液20μl注入液相色谱仪,其中杂质a、杂质d、杂质f、杂质g、杂质h、杂质k、杂质j、格列吡嗪、杂质e、杂质c、杂质i和杂质l依次出峰,各成分峰之间的分离度均应符合要求,保留时间分别为4.521min、5.620min、6.168min、8.421min、9.200min、10.650mon、11.599min、15.756min、17.282min、18.921min、21.821min、22.774min。取分离度溶液20ml注入高效液相色谱仪,记录色谱图。该方法具有下述不足:(1)该方法中声称可以“采用该方法可以实现对格列吡嗪和杂质a~l的定性检测和定量检测”,但是,对比文件1仅给出了11种杂质的测定色谱条件,其实施例和色谱图仅仅给出了添加杂质对照品的分离度实验色谱峰保留时间和色谱图,仅相当于方法学试验中的中的“专属性试验”,仅给出专属性实验图谱无法有效证明该方法用于定量测定格列吡嗪中杂质的准确性、可靠性和重现性。(2)该方法也未进一步明确是采取外标法、标准曲线法、还是自身主成分对照法进行测定,因此并不能确定其可准确对格列吡嗪中杂质进行定量测定。(3)该方法中化合物线性范围的下限和检测的定量限均较高,不适合于微量杂质的检测。技术实现要素:为了解决上述的技术问题,本发明提供了一种同时测定格列吡嗪类药物中8种化合物含量的检测方法,该方法能够准确定量测定格列吡嗪类药物中8种杂质。本发明是通过下述的技术方案来解决以上的技术问题的:一种同时测定格列吡嗪类药物中8种化合物含量的检测方法,所述测定方法为反相高效液相色谱法。所述格列吡嗪类药物为主成分为格列吡嗪的药物,所述主成分为格列吡嗪的药物包含格列吡嗪原料药、格列吡嗪合成中间体、格列吡嗪片、格列吡嗪胶囊、格列吡嗪缓释片、格列吡嗪分散片。上述的同时测定格列吡嗪类药物中8种化合物含量的检测方法中,所述8种化合物分别为化合物ⅰ:化合物ⅱ:化合物ⅲ:化合物ⅳ:化合物ⅴ:化合物ⅵ:化合物ⅶ:化合物ⅷ:上述的同时测定格列吡嗪类药物中8种化合物含量的检测方法中,所述反相高效液相色谱法包含如下步骤:(1)色谱条件;(2)供试品溶液制备;(3)混合对照品溶液制备;(4)测定法。所述色谱条件为:c18色谱柱;流动相a:0.05m磷酸二氢钾溶液,磷酸调节ph至ph3.4-3.6;流动相b:乙腈;流速:1.0ml·min-1;检测波长:223-227nmnm;柱温:30-40℃;进样量:20μl;梯度程序为:所述供试品溶液制备方法为:精密称取格列吡嗪原料药、格列吡嗪合成中间体、格列吡嗪片、格列吡嗪胶囊或格列吡嗪缓释片的细粉适量,约相当于格列吡嗪20mg,分别置50ml量瓶中,加稀释液(甲醇∶乙腈∶水=3∶8∶14)适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤,作为对照品溶液。所述混合对照品溶液制备制备方法为:精密称取化合物ⅰ、化合物ⅱ、化合物ⅲ、化合物ⅳ、化合物ⅴ、化合物ⅵ、化合物ⅶ、化合物ⅷ对照品适量,用稀释液溶解并定容至各含0.6μg/ml的溶液,作为混合对照品溶液;所述稀释液为由甲醇∶乙腈∶水按照3∶8∶14体积比比例配制。优选的,上述的同时测定格列吡嗪类药物中8种化合物含量的检测方法,包括如下步骤:色谱条件为:c18色谱柱;流动相a:0.05m磷酸二氢钾溶液,磷酸调节ph至3.5;流动相b:乙腈;流速:1.0ml·min-1;检测波长:225nm;柱温:35℃;进样量:20μl;梯度程序为:供试品溶液制备精密称取格列吡嗪原料药、格列吡嗪合成中间体、格列吡嗪片、格列吡嗪胶囊或格列吡嗪缓释片的细粉适量,约相当于格列吡嗪20mg,分别置50ml量瓶中,加稀释液(甲醇∶乙腈∶水=3∶8∶14)适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤,作为对照品溶液;混合对照品溶液制备精密称取化合物ⅰ、化合物ⅱ、化合物ⅲ、化合物ⅳ、化合物ⅴ、化合物ⅵ、化合物ⅶ、化合物ⅷ对照品适量,用稀释液溶解并定容至各含0.6μg/ml的溶液,作为混合对照品溶液;所述稀释液为由甲醇∶乙腈∶水按照3∶8∶14体积比比例配制。测定法精密量取供试品溶液和混合对照品溶液各20μl,分别注入液相色谱仪,记录色谱图,按外标法计算含量。上述的同时测定格列吡嗪类药物中8种化合物含量的检测方法,具体方法为:(1)色谱条件色谱柱:eclipsexdb-c18(250mm*4.6mm,5μm);流动相a:0.05m磷酸二氢钾溶液(磷酸调节ph至3.5);流动相b:乙腈;流速:1.0ml·min-1;检测波长:225nm;柱温:35℃;进样量:20μl;梯度程序(见表1)。表1梯度洗脱程序(2)检查方法供试品溶液制备精密称取格列吡嗪原料药、格列吡嗪合成中间体、格列吡嗪片、格列吡嗪胶囊或格列吡嗪缓释片的细粉适量(约相当于格列吡嗪20mg),分别置50ml量瓶中,加稀释液(甲醇∶乙腈∶水=3∶8∶14)适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤,作为对照品溶液。混合对照品溶液制备精密称取化合物ⅰ、化合物ⅱ、化合物ⅲ、化合物ⅳ、化合物ⅴ、化合物ⅵ、化合物ⅶ、化合物ⅷ对照品适量,用稀释液溶解并定容至各含0.6μg/ml的溶液,作为混合对照品溶液。测定法精密量取供试品溶液和混合对照品溶液各20μl,分别注入液相色谱仪,记录色谱图,按外标法计算含量。上述的测定方法在格列吡嗪原料或制剂生产中的用途。上述梯度洗脱设置中,所述洗脱时间指代该阶段洗脱所持续的时长,流动相的比例(va相:vb相,体积比)指代到达该洗脱时间终点时流动相的比例,其中阶段0指代流动相的起始比例,即:在该洗脱时间段内,流动相的比例由前一个时间段的终点值变至本时间段的终点值,如无特指,本发明中所有梯度设置的解释均遵从于此。杂质洗脱完毕后,流动相回调至初始流动相比例平衡,即可实现连续进样。流动相va相(%)、vb相(%)最终相加应为100%。优选的,流动相的比例在洗脱时间内为匀速改变。文献中关于格列吡嗪中杂质记载共有十三种,分别是上面表格中的记载的杂质a-杂质m。对于这些杂质,杂质b无紫外吸收,需采用气相色谱法检测;杂质m极性大,需采用反向离子对色谱法检测。通过控制格列吡嗪起始合成原料可以避免杂质j的产生,杂质k是一种二次降解产物,样品难生成,杂质l在合成中不易产生。在本发明的格列吡嗪类药物中8种化合物含量的检测方法方法开发前期,样品中杂质定性检测中,未检出杂质j、杂质k和杂质l。故本发明仅对样品中存在的其他8种杂质进行了对照品定性定量检测。本发明的有益效果:(1)本发明提供了一种同时测定格列吡嗪类药物中8种化合物含量的检测方法,该方法能够准确定量测定格列吡嗪类药物中8种杂质。(2)本发明的同时测定格列吡嗪类药物中8种化合物含量的检测方法,专属性试验、线性、范围、检出限、定量限、精密度、准确度、耐用性、溶液稳定性等具体试验,证明本方法(外标法)能够准确定量测定8种化合物的含量。(3)本申请中8种化合物线性范围的下限和检测的定量限均低于现有技术中要求的杂质配制浓度下限,证明本方法更适合微量杂质的检测。附图说明图1为本发明检测方法的专属性试验色谱图。说明8种化合物、格列吡嗪的分离度良好,分离度大于2.0(见专属性实验结果)。图2为本发明检测方法的混合对照品溶液典型色谱图。说明0.6μg/ml的混合杂质对照品溶液,各杂质色谱峰峰形良好,方法灵敏度高图3为本发明检测方法的供试品溶液典型色谱图。说明样品中的主成分和其他杂质不干扰8种化合物的测定。具体实施方式下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。具体情况如下:1、色谱条件色谱柱:eclipsexdb-c18(150mm*4.6mm,5μm);流动相a:0.05m磷酸二氢钾溶液(磷酸调节ph至3.5);流动相b:乙腈;流速:1.0ml·min-1;检测波长:225nm;柱温:35℃;进样量:20μl;梯度程序(见表1)。表1洗脱程序2、溶液配制稀释液:甲醇∶乙腈∶水=3∶8∶14化合物ⅰ对照品贮备溶液:精密称取化合物ⅰ对照品4.700mg置50ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅱ对照品贮备溶液:精密称取化合物ⅱ对照品4.201mg置50ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅲ对照品贮备溶液:精密称取化合物ⅲ对照品21.63mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅳ对照品贮备溶液:精密称取化合物ⅳ对照品22.25mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅴ对照品贮备溶液:精密称取化合物ⅴ对照品22.19mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅵ对照品贮备溶液:精密称取化合物ⅵ对照品19.78mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅶ对照品贮备溶液:精密称取化合物ⅶ对照品2.719mg置50ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。化合物ⅷ对照品贮备溶液:精密称取化合物ⅷ对照品20.18mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。格列吡嗪对照品贮备溶液:精密称取格列吡嗪对照品20.04mg置100ml量瓶中,加稀释液溶解并稀释至刻度,摇匀,2-8℃冷藏。专属性溶液:分别精密量取化合物ⅰ、化合物ⅱ和化合物ⅶ对照品贮备溶液各1ml和化合物ⅲ、化合物ⅳ、化合物ⅴ、化合物ⅵ、化合物ⅷ和格列吡嗪对照品贮备溶液各0.5ml,置同一10ml量瓶中,加稀释液稀释至刻度。混合对照品贮备溶液:分别精密量取化合物ⅰ、化合物ⅱ和化合物ⅶ对照品贮备溶液各10ml和化合物ⅲ、化合物ⅳ、化合物ⅴ、化合物ⅵ、化合物ⅷ对照品贮备溶液各5.00ml,置同一100ml量瓶中,加稀释液稀释至刻度。线性溶液1:精密量取20ml混合对照品贮备溶液,置50ml量瓶中加稀释液稀释至刻度。线性溶液2:精密量取10ml混合对照品贮备溶液,置50ml量瓶中加稀释液稀释至刻度。线性溶液3:精密量取10ml线性溶液1,置50ml量瓶中加稀释液稀释至刻度。线性溶液4:精密量取5ml线性溶液1,置50ml量瓶中,加稀释液稀释至刻度。线性溶液5:精密量取5ml线性溶液2,置50ml量瓶中,加稀释液稀释至刻度。线性溶液6:精密量取5ml线性溶液5,置10ml量瓶中,加稀释液稀释至刻度。线性溶液7:精密量取6ml线性溶液5,置25ml量瓶中,加稀释液稀释至刻度。3、专属性试验精密量取系统适应性溶液20μl,分别注入液相色谱仪,记录色谱图。结果:空白溶剂应无干扰,9种化合物之间的分离度均大于1.2,理论塔板数均大于5000,对称因子在0.5-2.0之间。表明方法专属性良好。表2专属性试验结果4、线性范围精密量取上述配制的不同浓度的线性溶液各20μl,分别注入液相色谱仪,记录色谱。结果化合物ⅰ分别在0.04512~3.76000μg/ml、化合物ⅱ在0.04033~3.36080μg/ml、化合物ⅲ在0.05176~4.31302μg/ml、化合物ⅳ在0.04913~4.09400μg/ml、化合物ⅴ在0.11073~4.42912μg/ml、化合物ⅵ在0.19068~3.81358μg/ml、化合物ⅶ在0.10876~2.17520μg/ml、化合物ⅷ在0.09989~3.99564μg/ml浓度范围内,浓度与峰面积线性关系良好。表3化合物ⅰ线性关系试验数据表4化合物ⅱ线性关系试验数据表5化合物ⅲ线性关系试验数据表6化合物ⅳ线性关系试验数据表7化合物ⅴ线性关系试验数据表8化合物ⅵ线性关系试验数据表9化合物ⅶ线性关系试验数据表10化合物ⅷ线性关系试验数据5、检测限和定量限用稀释液逐级稀释上述混合对照贮备溶液,进样,记录色谱图。以噪音的3倍作为检出限,以噪音的10倍作为定量限,结果见表。表11检测限和定量限结果6、精密度试验精密量取混合对品照贮备溶液3ml,置50ml量瓶中,加稀释液至刻度,作为混合对照品溶液。精密量取混合对照品溶液20μl,注入液相色谱仪连续进样6次,记录色谱。结果8种化合物峰面积rsd均小于2%,表明仪器精密度良好。表12系统精密度结果化合物溶液1溶液2溶液3溶液4溶液5溶液6平均值rsd%化合物ⅰ429604287743096429444323343269430630.4化合物ⅱ330313293433400331493344933876333061.1合物ⅲ375873685236706367613757237692371951.3化合物ⅳ409014080941775413174130742062413621.2化合物ⅴ304273032730353302993070530474304310.5化合物ⅵ275332717227643271552744727563274190.8化合物ⅶ122661193912339119651208712212121351.4化合物ⅷ193571921919382192741972719366193881.07、准确度供试品溶液:称取格列吡嗪原料20.31mg,置50ml量瓶中加稀释液适量,超声5min使溶解,加稀释液至刻度,得0.40mg/ml的溶液,25℃放置,现用现配。混合对照品溶液:精密量取混合对照品贮备溶液3.00ml,置50ml量瓶中,加稀释液至刻度,摇匀。准确度溶液:低浓度溶液:称取格列吡嗪原料20.69mg、20.59mg、20.32mg,分别置50ml量瓶中,各精密加入混合对照品贮备溶液1.00ml,加稀释液适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤。中浓度溶液:称取格列吡嗪原料19.92mg、20.04mg、20.07mg,分别置50ml量瓶中,各精密加入混合对照品贮备溶液3.00ml,加稀释液适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤。高浓度溶液:称取格列吡嗪原料19.91mg、20.23mg、20.05mg,分别置50ml量瓶中,各精密加入混合对照品贮备溶液6.00ml,加稀释液适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤。测定:精密量取上述溶液各20μl,分别注入液相色谱仪,记录色谱图。用外标法计算回收率。结论:8种化合物的平均回收均在90.0%-110.0%之间,且rsd均不大于5%,表明方法准确度良好。表13化合物ⅰ准确度试验结果表14化合物ⅱ准确度试验结果表15化合物ⅲ准确度试验结果表16化合物ⅳ准确度试验结果表17化合物ⅴ准确度试验结果表18化合物ⅵ准确度试验结果表19化合物ⅶ准确度试验结果表20化合物ⅷ准确度试验结果8、耐用性供试品溶液:取格列吡嗪原料约20mg,精密称定,置50ml量瓶中,加混合对照品贮备溶液3.00ml,加稀释液适量,超声5min使溶解,加稀释液稀释至刻度,摇匀,即得。25℃放置,现用现配。混合对照品溶液:精密量取混合对照品贮备溶液3ml,置50ml量瓶中,用溶剂稀释至刻度,摇匀。通过对色谱条件中流动相的ph值、测定波长、柱温、不同色谱柱进行微小的改变,重新进样测试,测定结果差异小,说明方法的耐用性良好。表21耐用性试验含量结果9、溶液稳定性试验取混合对照品贮备溶液3.00ml,置50ml量瓶中加稀释液稀释至刻度,摇匀,即得。精密量取20μl,分别在0、2、4、6、8、10、12小时取样进行检测,记录色谱图。结果8种化合物峰面积的rsd小于2.0%,表明溶液在12h内稳定。表22溶液稳定性试验结果系统适用性0h2h4h6h8h10h12hrsd化合物ⅰ848608448184876849658541885211851360.4化合物ⅱ339273350033787338153401434121346181.1化合物ⅲ368783681636962365993644836739369820.6化合物ⅳ419644194641338417924178441815422390.7化合物ⅴ305893072330252303343053330340302600.6化合物ⅵ282552768727978277372767727369281241.1化合物ⅶ123411240612153119571229812237120711.3化合物ⅷ193672000719602197331948719269193861.410、样品测定供试品溶液:取格列吡嗪原料药、格列吡嗪合成中间体、格列吡嗪片、格列吡嗪胶囊、格列吡嗪缓释片的细粉适量(约相当于格列吡嗪20mg),分别置50ml量瓶中,加稀释液适量,超声5min使溶解,加稀释液至刻度,摇匀,过滤,作为对照品溶液。混合对照品溶液:精密量取混合对照品贮备溶液3.00ml,置50ml量瓶中,加稀释液至刻度,摇匀。测定法:精密量取供试品溶液和混合对照品溶液各20μl,分别注入液相色谱仪,记录色谱图,按外标法计算含量。表23样品测定结果当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1