一种柑橘中调环酸的检测方法与流程
本发明涉及分析检测技术领域,尤其涉及一种柑橘中调环酸的检测方法。
背景技术:
调环酸钙(prohexadionecalcium)是一种新型的环己稀酮类植物生长调节剂,常用于农作物的生长调节,能有效地抑制作物节端生长,缩短茎秆长度,使茎秆粗壮、从而起到抗倒伏效果。调环酸钙的化学名称为3,5-二氧代-4-丙酰基环己烷羧酸钙,化学结构式为:
化学分子式c10h10cao5,分子量250.26。调环酸钙纯品为白色晶体,原药外观为米色或浅黄色无定形固体,易溶于水。调环酸钙是水溶性农药,难溶于有机溶剂。
目前,国内外对调环酸钙的研究主要集中在对作物的生长调控,对其在作物上的残留研究甚少。虽然调环酸钙属于低毒低残留农药,但其原药对胎鼠具有致畸毒性,因此调环酸钙在农作物中的残留量应引起人们的重视。gb2763-2019中调环酸钙的残留物定义为调环酸,以调环酸钙表示。
现有技术对调环酸钙的分析多采用高效液相色谱、uplc-ms/ms等。韦婕等报道了稻米中调环酸钙经乙腈(0.1%磷酸)两次提取,旋转蒸发后超纯水定容,未做净化处理直接上机检测。熊永等人使用乙腈、水和盐酸对花生等样品浸泡4h,振荡提取1h,未做净化处理直接检测。choi等建立了调环酸在白菜和苹果中的hplc-uvd。以上分析方法前处理耗时较长且净化不完全,而且国内外文献中未见到关于调环酸在柑橘样品中的残留检测方法。
技术实现要素:
有鉴于此,本发明的目的在于提供一种柑橘中调环酸的检测方法。本发明提供的检测方法前处理时间短、净化完全,结果准确可靠,可满足农药残留分析的要求,并可用于大量样品的快速检测。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种柑橘中调环酸的检测方法,包括以下步骤:
将待测柑橘样品和提取剂振荡混合后离心,取上清液作为提取液;所述提取剂为水;
所述提取液过混合型阴离子交换柱pax,得到吸附交换柱;
对所述吸附交换柱淋洗后,用甲酸水-甲醇溶液洗脱,收集洗脱液作为待测液;所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9;
采用高效液相色谱串联质谱仪对所述待测液进行检测,得到待测液中调环酸的色谱信息,基于调环酸的浓度-色谱信息标准曲线,得到待测柑橘样品中调环酸的含量。
优选地,所述高效液相色谱串联质谱仪的液相色谱条件包括:
色谱柱:watersacquity
流动相a为乙腈,流动相b为体积浓度为0.1%的甲酸水溶液;
洗脱程序为:
0~3min:5%流动相a;
3~4.5min:90%流动相a;
4.5~5.0min:5%流动相a;
流速:0.3ml/min;
进样量:1ul;
柱温:40℃;
所述高效液相色谱串联质谱仪的质谱条件包括:
离子源:esi-;
毛细管电压:2.5kv;
脱溶剂温度:600℃;
脱溶剂气流量:1000l/h;
检测方式:多重反应监测模式;母离子:211.00;锥孔电压30v;定量离子:123.00;定量离子的碰撞能量:14ev;定性离子:167.00,定性离子的碰撞能量:20ev。
优选地,所述待测柑橘样品与提取剂的用量比为5g:40ml。
优选地,所述混合型阴离子交换柱pax在使用前依次采用甲醇和水进行淋洗活化。
优选地,所述淋洗为依次进行的水淋洗和甲醇淋洗。
优选地,过混合型阴离子交换柱pax的提取液与水淋洗用水和甲醇淋洗用甲醇的体积比为8:5:10。
优选地,过混合型阴离子交换柱pax的提取液与甲酸水-甲醇溶液的体积比为8:5。
优选地,所述调环酸的浓度-色谱信息标准曲线的建立包括以下步骤:
将调环酸标准品用水配制成浓度分别为0.005、0.010、0.050、0.10和0.20mg/l的调环酸系列标准工作溶液;
采用高效液相色谱串联质谱仪对所述调环酸系列标准工作溶液进行检测,得到调环酸系列标准工作溶液的色谱信息;
将所述调环酸系列标准工作溶液的浓度和色谱信息进行线性拟合,得到所述调环酸的浓度-色谱信息标准曲线。
本发明提供了一种柑橘中调环酸的检测方法,采用水对待测样品进行振荡提取,然后用混合型阴离子交换柱pax和甲酸水-甲醇溶液对提取液进行净化处理,净化分离效果好。本方法的前处理(提取和净化)过程相对简单,回收率高,重复性好,净化分离效果好,而且最低检测浓度比较低,适合于调环酸在柑橘全果、果肉中的分析检测。实施例的数据表明:本发明提供的检测方法调环酸在0.005~0.20mg/l浓度范围内呈良好的线性关系;调环酸在柑橘全果中平均回收率为98~100%,相对标准偏差为0.9~5.2%;调环酸在柑橘果肉中平均回收率为102~104%,相对标准偏差为1.6~2.0%。本发明提供的检测方法操作简便、结果准确可靠、可满足农药残留分析的要求,并可用于大量样品的快速检测。
附图说明
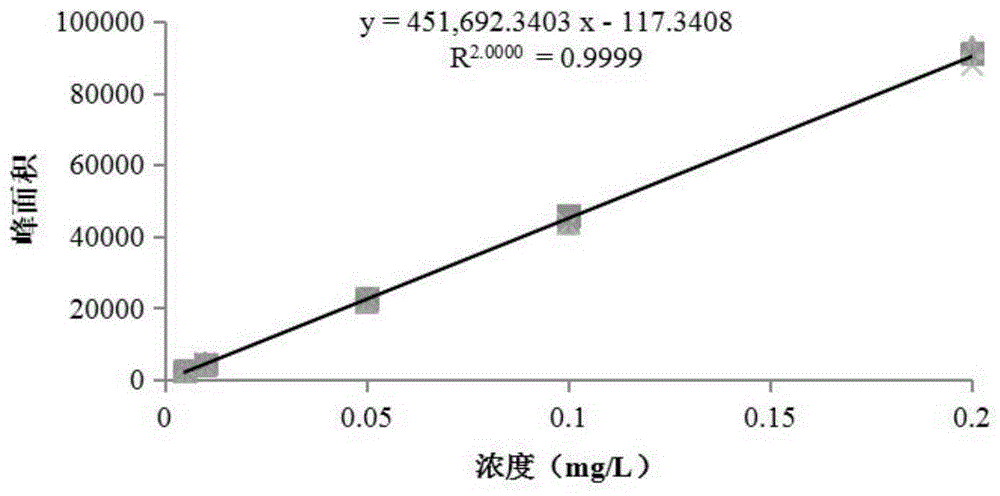
图1为调环酸的浓度-色谱信息标准曲线图。
具体实施方式
本发明提供了一种柑橘中调环酸的检测方法,包括以下步骤:
将待测柑橘样品和提取剂振荡混合后离心,取上清液作为提取液;
所述提取液过混合型阴离子交换柱pax,得到吸附交换柱;
对所述吸附交换柱进行淋洗后,用甲酸水-甲醇溶液洗脱,收集洗脱液作为待测液;所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9;
采用高效液相色谱串联质谱仪对所述待测液进行检测,得到待测液中调环酸的色谱信息,基于调环酸的浓度-色谱信息标准曲线,得到待测柑橘样品中调环酸的含量。
本发明将待测柑橘样品和提取剂振荡混合后离心,取上清液作为提取液。在本发明中,所述提取剂为水;所述待测柑橘样品与提取剂的用量比优选为5g:40ml;所述待测柑橘样品优选为柑橘全果或柑橘果肉。在本发明中,所述振荡的频率优选为2000~3000rpm,时间优选为25~35min。在本发明中,所述离心的转速优选为6500~7500rpm,时间优选为5min。在本发明中,优选再重复一次振荡混合后离心过程;每次添加的提取剂的量优选一样,即当待测柑橘样品的质量为5g时,每次提取剂的加入量为20ml,重复两次。
本发明采用提取剂水振荡,能够将待测柑橘样品中的调环酸高效提取出来。
得到提取液后,本发明将所述提取液过混合型阴离子交换柱pax,得到吸附交换柱。在本发明中,所述混合型阴离子交换柱pax在使用前优选依次采用甲醇和水进行淋洗活化;所述混合型阴离子交换柱pax优选为购自agelatechnologies公司的规格为500mg/6ml的混合型阴离子交换柱pax。
本发明将提取液过混合型阴离子交换柱pax,能够使提取液中离子化的调环酸吸附到混合型阴离子交换柱pax上。
得到吸附交换柱后,本发明对所述吸附交换柱进行淋洗后,用甲酸水-甲醇溶液洗脱,收集洗脱液作为待测液。在本发明中,所述淋洗优选为依次进行的水淋洗和甲醇淋洗;过混合型阴离子交换柱pax的提取液与水淋洗用水和甲醇淋洗用甲醇的体积比优选为8:5:10;淋洗后,本发明优选还包括将淋洗后的吸附柱挤干,本发明对所述挤干的方式不做具体限定。在本发明中,所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9;过混合型阴离子交换柱pax的提取液与甲酸水-甲醇溶液的体积比优选为8:5。
在本发明中,所述淋洗能够将吸附交换柱上的水溶性或者油溶性的杂质淋洗下来;然后用甲酸水-甲醇溶液将吸附交换柱上的调环酸洗脱下来,保证了洗脱液中调环酸的纯度。
得到待测液后,本发明采用高效液相色谱串联质谱仪对所述待测液进行检测,得到待测液中调环酸的色谱信息,基于调环酸的浓度-色谱信息标准曲线,得到待测柑橘样品中调环酸的含量。
在本发明中,当所述待测液的浓度较高,不满足高效液相色谱串联质谱仪的检测范围时,优选将所述待测液先进行稀释后再进行检测;本发明对所述稀释的参数不做具体限定,本领域技术人员根据实际情况进行选择即可。
在本发明中,所述高效液相色谱串联质谱仪的液相色谱条件包括:
色谱柱:watersacquity
流动相a为乙腈,流动相b为体积浓度为0.1%的甲酸水溶液;
洗脱程序为:
0~3min:5%流动相a;
3~4.5min:90%流动相a;
4.5~5.0min:5%流动相a;
流速:0.3ml/min;
进样量:1ul;
柱温:40℃;
所述高效液相色谱串联质谱仪的质谱条件包括:
离子源:esi-;
毛细管电压:2.5kv;
脱溶剂温度:600℃;
脱溶剂气流量:1000l/h;
检测方式:多重反应监测模式;母离子:211.00;锥孔电压30v;定量离子:123.00;定量离子的碰撞能量:14ev;定性离子:167.00,定性离子的碰撞能量:20ev。
在本发明中,所述调环酸的浓度-色谱信息标准曲线的建立优选包括以下步骤:
将调环酸标准品用水配制成浓度分别为0.005、0.010、0.050、0.10和0.20mg/l的调环酸系列标准工作溶液;
采用高效液相色谱串联质谱仪对所述调环酸系列标准工作溶液进行检测,得到调环酸系列标准工作溶液的色谱信息;
将所述调环酸系列标准工作溶液的浓度和色谱信息进行线性拟合,得到所述调环酸的浓度-色谱信息标准曲线。
本发明将调环酸标准品用水配制成浓度分别为0.005、0.010、0.050、0.10和0.20mg/l的调环酸系列标准工作溶液。本发明对所述调环酸系列标准工作溶液的配制方法不做具体限定,采用本领域技术人员公知的溶液配制方法即可。
得到调环酸系列标准工作溶液后,本发明采用高效液相色谱串联质谱仪对所述调环酸系列标准工作溶液进行检测,得到调环酸系列标准工作溶液的色谱信息。
在本发明中,所述高效液相色谱串联质谱仪的液相色谱条件和质谱条件优选与上述技术方案一致,在此不再赘述。
得到调环酸系列标准工作溶液的色谱信息后,本发明将所述调环酸系列标准工作溶液的浓度和色谱信息进行线性拟合,得到所述调环酸的浓度-色谱信息标准曲线。本发明对所述线性拟合的方式不做具体限定,采用本领域技术人员熟知的方式进行即可。
所述待测液中调环酸的色谱信息代入调环酸的浓度-色谱信息标准曲线后,直接得到的是待测液中调环酸的浓度;再优选基于公式1得到柑橘样品中调环酸的浓度:
公式1中,r为柑橘样品中调环酸的浓度,单位为mg/kg;c为待测液中调环酸的浓度,单位为mg/l;v为洗脱液甲酸水-甲醇溶液的体积,单位为ml;m为待测柑橘样品的质量,单位为g;f为过混合型阴离子交换柱pax的提取液与提取剂的体积比。
下面结合实施例对本发明提供的柑橘中调环酸的检测方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
以下实施例中待测液的制备方法均为:
将5g待测样品,置于50ml离心管中,加入20ml水振荡30min,7000rpm离心5min,重复上述步骤1次,合并上清液作为提取液;
分别用5ml甲醇、5ml水淋洗活化混合型阴离子交换柱pax(500mg/6ml);将8ml提取液上样,上样结束后分别用5ml水、10ml甲醇淋洗交换柱,挤干淋洗后的交换柱;再用5ml甲酸水-甲醇溶液(所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9)洗脱,收集洗脱液,过0.22μm有机相滤膜,作为待测液。
所用高效液相色谱串联质谱仪的液相色谱条件包括:
色谱柱:watersacquity
流动相a为乙腈,流动相b为体积浓度为0.1%的甲酸水溶液;
洗脱程序为:
0~3min:5%流动相a;
3~4.5min:90%流动相a;
4.5~5.0min:5%流动相a;
流速:0.3ml/min;
进样量:1ul;
柱温:40℃;
所述高效液相色谱串联质谱仪的质谱条件包括:
离子源:esi-;
毛细管电压:2.5kv;
脱溶剂温度:600℃;
脱溶剂气流量:1000l/h;
检测方式:多重反应监测模式;母离子:211.00;锥孔电压30v;定量离子:123.00;定量离子的碰撞能量:14ev;定性离子:167.00,定性离子的碰撞能量:20ev。
实施例1
调环酸的浓度-色谱信息标准曲线的建立:
取调环酸标准品,用水配制成浓度分别为0.005、0.010、0.050、0.10和0.20mg/l的调环酸系列标准工作溶液,在上述液相色谱和质谱条件下进行检测,得到所述调环酸系列标准工作溶液的色谱信息,结果如图1和表1所示。
表1调环酸的色谱信息
将所述调环酸系列标准工作溶液的浓度和色谱信息进行线性拟合,得到所述调环酸的浓度-色谱信息标准曲线,为y=451692.3403x-117.3408(r=0.9999),所得标准曲线如图1所示。
实施例2
方法的回收率和检出限
根据调环酸在高效液相色谱串联质谱仪上的响应情况,调环酸在柑橘全果待测样品中设定了3档添加浓度(0.05、0.5和1.0mg/kg),调环酸在柑橘果肉待测样品中设定了3档添加浓度(0.05、0.5和1.0mg/kg)每档添加浓度设5个重复。
按照上述的样品处理和检测参数,进行添加回收率试验。
得到柑橘全果、柑橘果肉样品中调环酸的添加回收率和相对标准偏差(rsds)如表2所示。
表2调环酸的添加回收率
从表2可知,调环酸在柑橘全果中添加浓度为0.05、0.5和1.0mg/kg时,平均回收率分别为98%、100%和100%,相对标准偏差分别为5.2%、1.5%和0.9%。调环酸在柑橘橘肉中添加浓度为0.05、0.5和1.0mg/kg时,平均回收率分别为104%、106%和102%,相对标准偏差分别为1.8%、1.6%和2.0%。高效液相色谱串联质谱仪对调环酸的最小检出量为5.00×10-12g。
根据调环酸在高效液相色谱串联质谱仪上的响应情况,以及柑橘全果、柑橘果肉和柑橘皮样品中调环酸最低添加浓度0.05mg/kg的样品在仪器上响应值均大于仪器的3倍信号噪音比,得出:调环酸在柑橘全果、果肉、皮样品中的最低检测浓度均为0.05mg/kg。由此说明:本发明提供的检测方法完全满足农药残留分析中对准确度、精密度和灵敏度的要求。
实施例3
称取5g(精确到0.01g)浙江柑橘全果样品,置于50ml离心管中,加入20ml水振荡30min,7000rpm离心5min,重复上述步骤1次,合并上清液作为提取液;
分别用5ml甲醇、5ml水淋洗活化混合型阴离子交换柱pax(500mg/6ml);将8ml提取液上样,上样结束后分别用5ml水、10ml甲醇淋洗吸附交换柱,然后挤干淋洗后的交换柱;再用5ml甲酸水-甲醇溶液(所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9)洗脱,收集洗脱液,过0.22μm有机相滤膜,得到柑橘全果待测液。
在上述液相色谱和质谱条件下进行检测,得到所述柑橘全果的调环酸色谱信息,峰面积为4481;
再基于调环酸的浓度-色谱信息标准曲线,为y=451692.3403x-117.3408,计算出待测液中调环酸的浓度为0.01mg/l;再结合公式1:
其中,c为0.01mg/l,v为5ml,m为5g,f为8/40;
得到柑橘全果中调环酸的含量为0.05mg/kg。
实施例4
称取5g(精确到0.01g)福建柑橘果肉样品,置于50ml离心管中,加入20ml水振荡30min,7000rpm离心5min,重复上述步骤1次,合并上清液作为提取液;
分别用5ml甲醇、5ml水淋洗活化混合型阴离子交换柱pax(500mg/6ml)小柱;将8ml提取液上样,上样结束后分别用5ml水、10ml甲醇淋洗吸附交换柱,然后挤干离心后的交换柱;再用5ml甲酸水-甲醇溶液(所述甲酸水-甲醇溶液由甲酸水溶液和甲醇混合而成,所述甲酸水溶液的体积浓度为10%,所述甲酸水溶液与甲醇的体积比为1:9)洗脱,收集洗脱液,过0.22μm有机相滤膜,得到柑橘果肉待测液。
在上述液相色谱和质谱条件下进行检测,得到所述柑橘果肉的调环酸色谱信息,峰面积为45772;
再基于调环酸的浓度-色谱信息标准曲线,y=451692.3403x-117.3408,得到待测液中调环酸的浓度为0.10mg/l;
再基于公式1:
其中,c为0.10mg/l,v为5ml,m为5g,f为8/40;
得到柑橘果肉中调环酸的含量为0.5mg/kg。
对比例1
与实施例3类似,区别仅在于将20ml水振荡,替换为20ml甲醇水溶液振荡,所述甲醇水溶液中甲醇和水的体积比为1:1。
对比例2
与实施例3类似,区别仅在于将20ml水振荡,替换为20ml乙腈振荡。
观察实施例3和对比例1和2可以看出:当使用甲醇水溶液或乙腈作为提取溶剂,调环酸的提取效率分别为70.8%和49.4%,提取率达不到要求;而水振荡提取效率为91.5%,提取效率能够达到要求。
提取效率(%)=(加标试样测定值-试样测定值)÷加标量×100%
对比例3
与实施例2类似,区别仅在于将混合型阴离子交换柱pax(500mg/6ml)替换为强阴离子交换柱sax(500mg/6ml)。
对比例4
与实施例2类似,区别仅在于将混合型阴离子交换柱pax(500mg/6ml)替换为florisil(500mg/6ml)。
对比例5
与实施例2类似,区别仅在于将混合型阴离子交换柱pax(500mg/6ml)替换为hlb(500mg/6ml)。
通过对比实施例2和对比例3~5可以看出:经过hlb、florisil净化柱后,柑橘全果中调环酸的回收率为59.4%和60.2%,回收率达不到要求且颜色杂质去除不净。相反,采用混合型阴离子交换柱pax和强阴离子交换柱sax进行净化,经过离子交换分配净化后,sax的回收率70.2%,pax净化后调环酸的回收率为97.3%,回收率能够达到要求;而且净化后液体为无色,去除杂质的效果很好。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!