液态渔用中草药制剂中抗生素类药物的测定方法与流程
1.本发明属于抗生素类药物测定领域,尤其涉及一种液态渔用中草药制剂中抗生素类药物的测定方法。
背景技术:
2.渔用中草药制剂具有天然、高效、毒副作用少、增强免疫功能,无抗药性等优点,兼有药用和营养双重作用,在水产养殖中的应用越来越广泛。与化学药物相比,中草药药性温和、药效反应时间较长,主要适用于预防水产养殖动物的疾病。为了提高渔用中草药治疗水产养殖动物疾病的效果,渔用中草药制剂中添加一定量的抗生素,有利于治疗水产养殖动物疾病。但是,过量抗生素和非法添加的抗生素则会使其在水产养殖动物体内的残留,造成水产养殖动物自身连同其食用者产生耐药性,从而间接影响人体健康。
3.喹诺酮类和磺胺类抗生素是一类人工合成的广谱类抗生素药物,被广泛应用于水产养殖行业。根据gb31650-2019食品中兽药最大残留限量规定,鱼肌肉和皮中该类药物的最大残留限量为:达氟沙星100μg/kg、二氟沙星300μg/kg、恩诺沙星(恩诺沙星与环丙沙星之和)100μg/kg、氟甲喹500μg/kg、噁喹酸100μg/kg、沙拉沙星30μg/kg、磺胺总量100μg/kg。而根据农业部公告第2292号规定,洛美沙星、培氟沙星、氧氟沙星、诺氟沙星属于停用药,禁止在养殖中使用。为了能更好地规范渔用中草药制剂中抗生素类药物的生产与使用,排查中草药制剂存在的潜在风险隐患,建立渔用中草药制剂中抗生素类药物测定的检测分析方法显得尤为重要。
4.渔用中草药制剂不仅基质复杂,且部分市售药物为液态,需要进一步的富集除杂。传统的液液萃取法、固相萃取法存在试剂消耗量大、操作冗繁的缺点,不适用于快速分析。且现有的国家标准均不适用于液态渔用中草药制剂中抗生素的残留测定,无法有效监控其残留状况。
技术实现要素:
5.本发明的目的在于提出一种液态渔用中草药制剂中抗生素类药物的测定方法,采用分散液液微萃取结合液质联用技术,同时测定液态渔用中草药制剂中的喹诺酮类、磺胺类药的残留。
6.为达此目的,本发明采用以下技术方案:
7.本发明提供的液态渔用中草药制剂中抗生素类药物的测定方法,包括以下步骤:s1:将内标混合物加入样液中涡旋0.4-0.6min,静置4-6min后,加入乙腈,涡旋0.8-1.2min,得混合溶液,s2:将混合液体积的1/4-1/6移取出,依次往移取出的混合液加入磷酸盐缓冲溶液和三氯甲烷,涡旋0.8-1.2min,形成磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系,s3:将磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系离心2.5-3.5min,抽取1.0-1.5ml沉淀相,以38-42℃的氮气吹干,吹干后加入定容液,然后进行超声溶解,超声溶解后经滤膜过滤后经高效液相色谱-串联质谱仪测定。
8.优选地,磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系中,磷酸盐缓冲溶液、三氯甲烷、乙腈乳浊液的体积比为9.8-10.2:1.4-1.6:1.5-1.8。
9.优选地,步骤s1具体包括以下步骤:称取1.0ml样液于离心管中,加入0.5ml内标混合液,以2000r/min的转速涡旋0.5min,静置5min后,加入8.5ml乙腈,以2500r/min的转速涡旋1min,得10ml混合溶液。
10.优选地,步骤s2具体包括以下步骤:从10ml混合溶液中移取出2ml混合液于离心管中,依次加入9.8ml磷酸盐缓冲溶液和1.5ml三氯甲烷,以2500r/min的转速涡旋1min,形成磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系。
11.优选地,步骤s3具体包括以下步骤:将磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系以2000r/min的转速离心3min,用微量进样器抽取1.0-1.5ml沉淀相,以40℃的氮气吹干,吹干后加入1.0ml定容液,然后进行超声溶解,超声溶解后经0.22μm滤膜过滤后经高效液相色谱-串联质谱仪测定。
12.优选地,磷酸盐缓冲溶液的ph值为7.0,浓度为0.05mol/l。
13.优选地,内标混合物为诺氟沙-d5、恩诺沙星-d5、磺胺邻二甲氧嘧啶-d3和磺胺间二甲氧嘧啶-d6的甲醇溶液,浓度为0.5μg/ml。
14.优选地,步骤s3中,色谱条件为:采用规格为2.1mm
×
150mm
×
5μm的ultimate xb-c
18
色谱柱,柱温35℃,流速0.25ml/min,进样量10μl;流动相:a为0.002mol/l乙酸铵-0.1%甲酸水溶液,b为乙腈;洗脱梯度:0-5.5min,15%-20%b;15.5-16min,20%-40%b;16-23min,40%b;23-23.5min,40%-15%b;23.5-28min,15%b。
15.优选地,步骤s3中,质谱条件为:采用电喷雾离子源,正离子检测模式,喷雾电压:3500v,鞘气压力241kpa,辅助气压力:2l/min,离子传输毛细管温度:350℃,选择反应监测模式,q1半峰宽:0.7u,q3半峰宽:0.7u,碰撞气压力:氩气,0.2pa。
16.本发明的有益效果为:
17.1、考虑乙腈可作为同时作为提取剂和分散剂的特性,先采用乙腈直接提取富集中草药制剂中抗生素,再加入磷酸盐缓冲液,然后借由乙腈与水互溶的特点及作为分散剂的特性,溶于乙腈的抗生素进入磷酸盐缓冲液。随后通过萃取剂三氯甲烷的加入,则顺利形成磷酸盐缓冲液/三氯甲烷/乙腈乳浊体系,快速将抗生素提取至三氯甲烷,实现了快速dllme。同时,磷酸盐缓冲液稀释部分提取液的方案,也解决后续dllme两相无法分层及色素的基质抑制效应问题。
18.2、采用分散液液微萃取技术与液质联用技术结合,可快速有效地监测液态渔用中草药制剂中抗生素类药物的含量。
19.3、通过对磷酸盐缓冲溶液、三氯甲烷、乙腈的体积配比优化、以及乳浊体系ph值的调控,提高目标分析物回收率,获得较高的萃取效率。
20.4、萃取时间为1.0min,提高萃取方法的重现性。
21.5、内标的引入,则改善了由于磷酸盐缓冲液稀释引起的方法灵敏度下降的情况。
附图说明
22.图1是本发明实施例的标准品选择反应监测离子流色谱图。
23.图2是本发明实施例的ph值对目标分析物回收率的影响图。
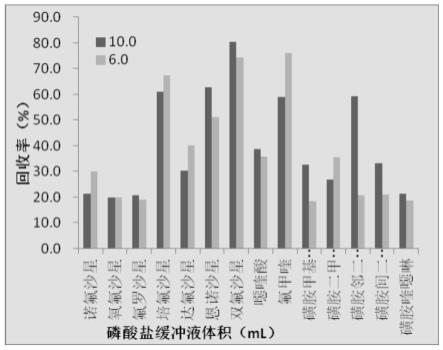
24.图3是本发明实施例的磷酸盐缓冲液体积对目标分析物回收率的影响图。
25.图4是本发明实施例的萃取剂对目标分析物回收率的影响图。
26.图5是本发明实施例的萃取剂体积对目标分析物回收率的影响图。
27.图6是本发明实施例的分散剂体积对目标分析物回收率的影响图。
具体实施方式
28.现结合附图和具体实施方式对本发明进一步说明。
29.仪器与试剂:tsq quantum ultra高效液相色谱-串联质谱仪(美国thermo fisher公司),配电喷雾离子源;ab204-e型、pl203型电子分析天平(mettler toledo公司);3-5w小型台式低速离心机(湖南恒诺仪器设备有限公司);ms3型旋涡混合器(德国ika公司);hsc-24b水浴氮吹仪(天津市恒奥科技发展有限公司);milli-q型超纯水仪(美国millipore公司);0.22μm尼龙微孔滤膜(天津市津腾实验设备有限公司)。
30.诺氟沙星(norfloxacin)、氧氟沙星(ofloxacin)、氟罗沙星(fleroxacin)、甲磺酸达氟沙星(danofloxacin mesylate)、恩诺沙星(enrofloxacin)、盐酸二氟沙星(difloxacin hydrochloride)、噁喹酸(oxolinic acid)、氟甲喹(flumequine)、磺胺甲基嘧啶(sulfamerazine)、磺胺二甲基嘧啶(sulfamethazine)、磺胺邻二甲氧嘧啶(sulfadoxine)、磺胺间二甲氧嘧啶(sulfadimethoxine)、磺胺喹噁啉(sulfachinoxalin),标准品纯度大于97%;甲磺酸培氟沙星(pefloxacin methanesulfonate),标准品纯度大于92%;购于德国dr.ehrenstorfer gmbh公司。上述标准品均用甲醇配制成100μg/ml的标准储备液,-20℃下避光保存(称取标准品的质量是按纯度修正过的质量)。再用甲醇配制成1μg/ml的14种抗生素标准混合液,-20℃下避光保存。氘代诺氟沙星(norfloxacin-d5)、氘代恩诺沙星(enrofloxacine-d5)、氘代磺胺邻二甲氧嘧啶(sulfadoxine-d3)、氘代磺胺间二甲氧嘧啶(sulfadimethoxine-d6),标准品纯度5μg/ml、扩展不确定度0.2μg/ml;购于农业农村部农产品质量标准研究中心。用甲醇配制成0.5μg/ml标准内标混合液,-20℃下避光保存。
31.甲醇、乙腈、甲酸均为色谱纯,购自美国tidea公司;其他试剂均为分析纯,购自国药集团化学试剂有限公司;实验用水为milli-q超纯水。0.05mol/l磷酸盐缓冲溶液(ph7.0):取磷酸二氢钠1.56g,加入氢氧化钠溶液79ml,混匀,用水稀释至200ml。定容液:取乙腈15ml,取0.1%甲酸水溶液(含0.002mol/l乙酸铵)85ml,混合均匀。石墨化碳黑购置天津博纳艾杰尔科技有限公司。
32.本实施例中提供的液态渔用中草药制剂中抗生素类药物的测定方法,包括以下步骤:
33.s1:称取1.0ml样液于15ml玻璃离心管中,加入0.5μg/ml标准内标混合液(诺氟沙-d5、恩诺沙星-d5、磺胺邻二甲氧嘧啶-d3和磺胺间二甲氧嘧啶-d6的甲醇溶液)0.5ml,以2000r/min的转速涡旋0.5min。静置5min后,加入8.5ml乙腈,以2500r/min的转速涡旋1min,得10ml混合溶液。
34.s2:从10ml混合溶液中移取出2ml混合液于15ml玻璃离心管中,依次加入9.8ml磷酸盐缓冲溶液和1.5ml三氯甲烷,以2500r/min的转速涡旋1min,形成磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系。
35.s3:将磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系以2000r/min的转速离心3min,用微量进样器抽取1.0ml沉淀相,以40℃的氮气吹干,吹干后加入1.0ml定容液,然后进行超声溶解,超声溶解后经0.22μm滤膜过滤后经高效液相色谱-串联质谱仪测定。
36.本实施例中,磷酸盐缓冲溶液、三氯甲烷、乙腈乳浊液的体积比为9.8:1.5:1.7。
37.其中,磷酸盐缓冲溶液的ph值为7.0,浓度为0.05mol/l。由于磷酸盐缓冲溶液的ph值为7.0,三氯甲烷为中性,乙腈为极弱碱性,因此三者组成的磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系为中性。喹诺酮类药物存在羧基和哌嗪基,在不同ph值条件下,可形成正离子、负离子和中性分子。当喹诺酮类药物以中性分子状态存在时,其在有机相中的分配系数提高,萃取效率增大。采用呈中性的磷酸盐缓冲溶液/三氯甲烷/乙腈乳浊液体系的萃取效率更高。
38.其中,步骤s3中,色谱条件为:采用规格为2.1mm
×
150mm
×
5μm的ultimate xb-c
18
色谱柱,柱温35℃,流速0.25ml/min,进样量10μl;流动相:a为0.002mol/l乙酸铵-0.1%甲酸水溶液,b为乙腈;洗脱梯度:0-5.5min,15%-20%b;15.5-16min,20%-40%b;16-23min,40%b;23-23.5min,40%-15%b;23.5-28min,15%b。
39.其中,步骤s3中,质谱条件为:采用电喷雾离子源,正离子检测模式,喷雾电压:3500v,鞘气压力241kpa,辅助气压力:2l/min,离子传输毛细管温度:350℃,选择反应监测模式,q1半峰宽:0.7u,q3半峰宽:0.7u,碰撞气压力:氩气,0.2pa。母离子、子离子和碰撞能量列见表1。标准品选择反应监测离子流色谱图示见图1。
[0040][0041][0042]
表1选择反应监测母离子、子离子和碰撞能量
[0043]
注:*为定量碎片离子。
[0044]
提取方法优化:
[0045]
市售的中草药制剂大多为悬浊液,实际生产使用过程中,采用的方法为直接泼洒法。因此,悬浊液中的固体和液体所含的抗生素均会对养殖环境造成影响。为了充分提取出其所含的抗生素,与常规水样的前处理不同,市售的中草药制剂不能过膜除去固体后再进行提取,应采取直接提取法。
[0046]
首先选取提取效率最高的乙腈作为直接提取剂,但由于乙腈和水互溶,后续无法浓缩至干,无法进一步富集中草药制剂中的抗生素。于是改为直接采用dllme,但由于中草药制剂色素含量高,且液体中含有悬浮状固体,因此两相分层不明显,稳定性较差。考虑乙
腈可作为同时作为提取剂和分散剂的特性,先采用乙腈直接提取富集中草药制剂中抗生素,再加入磷酸盐缓冲液,然后借由乙腈与水互溶的特点及作为分散剂的特性,溶于乙腈的抗生素进入磷酸盐缓冲液。随后通过萃取剂三氯甲烷的加入,则顺利形成磷酸盐缓冲液/三氯甲烷/乙腈乳浊体系,快速将抗生素提取至三氯甲烷,实现了快速dllme。同时,磷酸盐缓冲液稀释部分提取液的方案,也解决后续dllme两相无法分层及色素的基质抑制效应问题。而内标的引入,则改善了由于磷酸盐缓冲液稀释引起的方法灵敏度下降的情况。
[0047]
分散液液微萃取条件优化:
[0048]
确定提取方法后,采用10ml加标100ng的磷酸盐缓冲液进行dllme各种条件的优化,重复3次取平均值计算。
[0049]
ph值:
[0050]
当目标分析物在非解离状态下,dllme的萃取结果较理想。因此,溶液ph值会改变抗生素的存在状态,进而影响其回收率。喹诺酮类药物存在羧基和哌嗪基,在不同ph值条件下,可形成正离子、负离子和中性分子。当喹诺酮类药物以中性分子状态存在时,其在有机相中的分配系数提高,萃取效率增大。同样磺胺类药物在酸性或碱性的溶液中有较大溶解度,不利于后续的有机萃取。如图2的结果表明,在0.8ml乙腈分散剂和0.6ml三氯甲烷萃取剂条件下,大多数喹诺酮和磺胺类抗生素药物与三氯甲烷萃取剂的分配系数最高,表现较高的萃取效率。因此,选用磷酸盐缓冲溶液的ph为中性(7.0)。
[0051]
磷酸盐缓冲液体积的影响:
[0052]
磷酸盐缓冲液是本方法目标分析物的载体。磷酸盐缓冲液体积与分散剂、萃取剂的体积比对萃取结果有着重要的影响。考察了10.0ml、6.0ml磷酸盐缓冲液样对目标分析物回收率的影响。如图3所示,结果表明,在0.8ml乙腈分散剂、0.6ml三氯甲烷萃取剂、磷酸盐缓冲液ph为中性条件下,10.0ml的磷酸盐缓冲液体积可以获得较优的萃取效率。
[0053]
萃取剂及体积的优化:
[0054]
萃取剂是影响dllme萃取效率的重要因素,其密度、溶解度、粘度和萃取容量是影响萃取效果和重现性的重要因素。(1)萃取剂的密度大于或者小于水和分散剂密度,易于从混合溶液中获得;(2)萃取剂要易与分散剂混溶而不溶于样液;(3)能形成稳定的两相系统;(4)萃取剂对目标分析物具有较好的萃取能力。分别选取二氯甲烷、三氯甲烷作为萃取剂,在中性样液条件下,以0.8ml乙腈作为分散剂考察萃取效率。如图4所示,结果表明,二氯化碳与分散剂混合后,液滴损失严重,萃取效率低且重复性差;而三氯甲烷对目标分析物的萃取效率较佳。
[0055]
在同等条件下,考察了萃取剂体积对目标分析物回收率的影响。如图5所示,结果表明,目标分析物的回收率随着萃取剂体积的增加而增加,但当体积增至1.5ml时,目标分析物回收率基本稳定不变。这说明随着三氯甲烷用量的增大,进入有机相的目标分析物增多。因此,最终选择1.5ml的三氯甲烷最为最佳的萃取条件。
[0056]
分散剂体积的优化:
[0057]
分散剂乙腈用量直接影响磷酸盐缓冲液/三氯甲烷/乙腈乳浊体系的形成,影响萃取剂在水中的分散程度,从而影响目标分析物的回收率。考察了乙腈体积(0.6、0.8、1.2、1.4、1.6、1.8ml)对目标分析物回收率的影响。如图6所示,结果表明,目标分析物回收率随乙腈体积的增加而增加,当乙腈体积增至1.6ml时,目标分析物回收率达到最大值。而当体
积增至1.8ml时,目标分析物回收率无明显变化。因此选择分散剂乙腈的最佳体积为1.6ml。
[0058]
萃取时间的影响:
[0059]
考察了不同萃取时间(0.5、1.0、2.0、3.0min)对萃取效率的影响。结果表明,涡旋时间的长短对萃取效率影响不大,但可以提高萃取方法的重现性。综合考虑上述因素后,选择萃取时间为1.0min。
[0060]
标准曲线、线性范围、检出限和定量限:
[0061]
为了消除基质效应带来的定量偏差,实验采用基质匹配标准曲线法。移取适量混合标准溶液,用空白样品提取液分别配制成不同质量浓度基质标准溶液,目标物浓度分别为10,50,100,200,500,1000ng/ml。以各组分浓度与其色谱峰面积进行线性回归,呈良好线性关系,相关系数均大于0.99。以10倍信噪比(s/n)计算定量限(limit of quantitation,loq),具体数值列见表2。
[0062]
方法准确度和精密度:
[0063]
以市售渔用中草药制剂为研究对象进行标准添加实验,分别以低、中、高三个添加水平进行加标回收实验、每个浓度水平做6次平行实验,考察方法的准确度及精密度。表2结果表明:平均回收率在60.0%-119.7%,相对标准偏差1.0%-14.7%。30天内,1μg/l加标浓度下进行5次标准添加实验,考察方法日间精密度,相对标准偏差3.0%-12.3%(见
[0064]
表2)。方法的精密度和准确度均能满足药物残留监测需求。
[0065][0066]
表2方法准确度和精密度测定结果
[0067]
分散液液微萃取(dispersive liquid-liquid microextraction,dllme)技术具有操作简单、快速、灵敏度高、环境友好型、价格低廉等优点,与液质联用技术结合,可快速有效地监测液态渔用中草药制剂中抗生素类药物的含量。
[0068]
本实施例以渔用中草药制剂为研究对象,分别考察了萃取剂及体积、分散剂及体积、ph值对目标抗生素回收率的影响,建立了分散液液微萃取结合液质联用技术同时测定渔用中草药制剂中的喹诺酮类、磺胺类药残留的快速分析方法。能加快推进水产养殖业绿
色高质量发展,并为水产品质量安全监管工作提供技术支撑。
[0069]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解;其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1