诺氟沙星的检测方法以及荧光检测材料的制备方法与流程
1.本发明涉及诺氟沙星的检测的技术领域,具体而言,涉及诺氟沙星的检测方法以及荧光检测材料的制备方法。
背景技术:
2.诺氟沙星(norfloxacin,nor)是氟喹诺酮类药物中的一种,广泛应用于医学。nor对革兰氏阳性、革兰氏阴性菌和支原体引起感染,如肺部、尿液和消化系统感染。近年来,氟喹诺酮类药物在环境中积累,对生态系统和人类健康构成威胁,对生态系统和人类健康构成威胁,由此开发一种灵敏、有效的检测环境中nor的方法至关重要。到目前为止,关于检测nor的分析方法报道的很多,如电化学分析、化学发光、高效液相色谱(hplc)、紫外可见分光光度法、滴度法等。然而,这些分析方法在实际应用时受到限制,例如,需要加热或提取步骤,涉及昂贵的试剂和繁琐的样品预处理,显示出狭窄的线性响应范围和时间消耗等。因此,有必要开发一种快速、简单、低成本的有效测定nor的方法。
3.荧光光谱法因具有操作简单、检测限低、响应时间快、特异性和灵敏度高、实用性强等诸多优点而备受关注。碳点(cds)是一类特殊的荧光纳米材料,具有明亮的荧光、高光稳定性、良好的生物相容性和优异的水溶性等特点。因此,将碳点作为荧光探针并应用于水中诺氟沙星的检测具有较高的可行性。但是申请人在实践中发现,因荧光变化程度小、特异性差(尤其是对抗生素药物的识别)以及抗干扰能力较差,单纯的碳点并不能应用于检测水中的微量诺氟沙星。
技术实现要素:
4.本发明的主要目的在于提供诺氟沙星的检测方法以及荧光检测材料的制备方法,以解决现有技术中荧光变化程度不足、特异性差以及抗干扰能力较差的技术问题。
5.为了实现上述目的,根据本发明的第一个方面,提供了诺氟沙星的检测方法,技术方案如下:
6.诺氟沙星的检测方法,采用n,s-cds,所述n,s-cds为氮和硫共掺杂的碳点;检测待测液的第一荧光强度以及待测液和n,s-cds的混合物的第二荧光强度,根据第二荧光强度和第一荧光强度的比值与诺氟沙星浓度的线性关系,换算得到待测液中诺氟沙星的浓度。
7.作为本发明第一方面的进一步改进,所述线性关系为:y=0.01709x+1.01243,r2=0.997,x为诺氟沙星浓度,x为0.5~50μmol/l,y为第二荧光强度和第一荧光强度的比值。
8.作为本发明第一方面的进一步改进,测试环境的ph为2~6;待测液和n,s-cds的反应温度为20~30℃;待测液和n,s-cds的反应时间为1~5min。
9.作为本发明第一方面的进一步改进,n,s-cds的xps全能谱图在400.1ev、231.1ev和163.81ev处具有特征峰;n,s-cds在n1s的xps光谱在398.8ev和400.9ev处具有特征峰;n,s-cds在s2p的xps光谱在164.2ev和163.2ev处具有特征峰。
10.作为本发明第一方面的进一步改进,n,s-cds的firt光谱在3432cm-1
、3180cm-1
、
1581cm-1
、1405cm-1
、1250cm-1
和615cm-1
处具有特征峰。
11.作为本发明第一方面的进一步改进,n,s-cds的制备包括以下步骤:对柠檬酸铵和l-半胱氨酸的混合液进行热处理和纯化处理,即得到n,s-cds。
12.为了实现上述目的,根据本发明的第二个方面,提供了用于检测诺氟沙星的荧光检测材料的制备方法,技术方案如下:
13.用于检测诺氟沙星的荧光检测材料的制备方法,包括以下步骤:
14.(1)获取包括柠檬酸铵和l-半胱氨酸的混合液;
15.(2)对混合液进行热处理;
16.(3)将热处理产物分散于水中,然后进行离心处理;
17.(4)对离心处理得到的上清液进行透析处理,即得到用于检测诺氟沙星的n,s-cds水溶液。
18.作为本发明第二方面的进一步改进,所述混合液中柠檬酸铵和l-半胱氨酸的质量比值为2~2.4,所述混合液按照每10ml水中加入0.8~1.2gl-半胱氨酸的配比配制得到。
19.作为本发明第二方面的进一步改进,热处理温度为170~190℃,时间为60~90min;透析处理采用截留分子量为1000da的透析袋,透析时间为18~30h。
20.作为本发明第二方面的进一步改进,还包括在热处理之前对混合液进行超声处理;还包括对离心处理得到的上清液进行过滤处理。
21.首先,本发明的诺氟沙星的检测方法利用氮和硫共掺杂改性的碳点对诺氟沙星进行荧光检测,不仅工艺简单,操作方便,而且荧光变化程度高,线性范围宽、检测限低,能够特异性识别诺氟沙星,且抗干扰能力强,有效解决了水中诺氟沙星难检测的技术问题。其次,本发明的氮和硫共掺杂改性的碳点的制备工艺简单可控,生产成本低。由此可见,本发明的诺氟沙星的检测方法以及荧光检测材料的制备方法的实用性强,非常适合于推广使用。
22.下面结合附图和具体实施方式对本发明做进一步的说明。本发明附加的方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
23.构成本发明的一部分的附图用来辅助对本发明的理解,附图中所提供的内容及其在本发明中有关的说明可用于解释本发明,但不构成对本发明的不当限定。在附图中:
24.图1为n,s-cds水溶液的f
1-f0随柠檬酸铵和l-半胱氨酸的质量比的变化曲线。
25.图2为n,s-cds水溶液的f
1-f0随热处理温度的变化曲线。
26.图3为n,s-cds水溶液的f
1-f0随热处理时间的变化曲线。
27.图4为n,s-cds粉末的tem照片和hr-tem照片(插图)。
28.图5为最佳制备工艺参数下所得n,s-cds的粒径分布图。
29.图6为n,s-cds粉末的傅里叶变换红外光谱。
30.图7为n,s-cds粉末的x射线光电子能谱全能谱图。
31.图8为n,s-cds粉末在c1s的xps光谱。
32.图9为n,s-cds粉末在n1s的xps光谱。
33.图10为n,s-cds粉末在o1s的xps光谱。
34.图11为n,s-cds粉末在s2p的xps光谱。
35.图12为n,s-cds水溶液以及n,s-cds+nor(n,s-cds水溶液和诺氟沙星溶液的混合物)的荧光寿命图。
36.图13为n,s-cds水溶液的f
1-f0随测试环境的ph的变化曲线。
37.图14为n,s-cds水溶液的f
1-f0随反应温度的变化曲线。
38.图15为n,s-cds水溶液的f
1-f0随反应时间的变化曲线。
39.图16为n,s-cds水溶液检测不同抗生素的效果对比图。
40.图17为n,s-cds水溶液的抗干扰性能的测试结果。
41.图18为n,s-cds水溶液测试不同浓度的诺氟沙星溶液的荧光谱图。
42.图19为n,s-cds水溶液的f1/f0与诺氟沙星浓度之间的线性拟合曲线。
具体实施方式
43.下面结合附图对本发明进行清楚、完整的说明。本领域普通技术人员在基于这些说明的情况下将能够实现本发明。在结合附图对本发明进行说明前,需要特别指出的是:
44.本发明中在包括下述说明在内的各部分中所提供的技术方案和技术特征,在不冲突的情况下,这些技术方案和技术特征可以相互组合。
45.此外,下述说明中涉及到的本发明的实施例通常仅是本发明一部分的实施例,而不是全部的实施例。因此,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
46.关于本发明中术语和单位。本发明的说明书和权利要求书及有关的部分中的术语“包括”、“具有”以及它们的任何变形,意图在于覆盖不排他的包含。
47.本发明的用于检测诺氟沙星(nor)的荧光检测材料的制备方法的实施例为包括以下步骤:
48.(1)获取包括柠檬酸铵和l-半胱氨酸的混合液;
49.(2)对混合液进行热处理;
50.(3)将热处理产物分散于水中,然后进行离心处理;
51.(4)对离心处理得到的上清液进行透析处理,即得到用于检测诺氟沙星的荧光检测材料(即n,s-cds水溶液)。
52.其中,所述混合液中柠檬酸铵和l-半胱氨酸的质量比值为2~2.4,可以但是不限于取值为2、2.1、2.2、2.3、2.4中的任意一个。
53.所述混合液按照每10ml水中加入0.8~1.2gl-半胱氨酸的配比配制得到,可以但是不限于取值为0.8g、0.9g、1g、1.1g、1.2g中的任意一个。
54.为了提升碳点粒度的均匀性,还包括在热处理之前对混合液进行超声处理直至形成澄清透明的混合液。
55.热处理温度为170~190℃,可以但是不限于取值为170℃、175℃、180℃、185℃、190℃中的任意一个,热处理时间为60~90min,可以但是不限于取值为60min、70min、80min、90min中的任意一个。
56.透析处理采用截留分子量为1000da的透析袋,透析时间为18~30h,可以但是不限
于取值为18h、20h、22h、24h、26h、28h、30h中的任意一个。
57.为了提升透析的效率,还包括对离心处理得到的上清液进行过滤处理,过滤处理优选采用孔径为0.2~0.25μm的滤针进行过滤,孔径可以但是不限于取值为0.2μm、0.21μm、0.22μm、0.23μm、0.24μm、0.25μm中的任意一个。
58.所得n,s-cds水溶液经冷冻干燥后可以得到n,s-cds粉末,从而求得n,s-cds水溶液中n,s-cds的浓度。经验证,n,s-cds粉末的xps全能谱图在400.1ev、231.1ev和163.81ev处具有特征峰;n,s-cds粉末在n1s的xps光谱在398.8ev和400.9ev处具有特征峰;n,s-cds粉末在s2p的xps光谱在164.2ev和163.2ev处具有特征峰。n,s-cds的firt光谱在3432cm-1
、3180cm-1
、1581cm-1
、1405cm-1
、1250cm-1
和615cm-1
处具有特征峰。
59.本发明的诺氟沙星的检测方法的实施例为采用n,s-cds,所述n,s-cds为氮和硫共掺杂的碳点,优选采用上述的制备方法制备得到的n,s-cds粉末或n,s-cds水溶液;使用时,检测待测液的第一荧光强度以及待测液和n,s-cds水溶液的混合物的第二荧光强度,根据第二荧光强度和第一荧光强度的比值与诺氟沙星浓度的线性关系,换算得到待测液中诺氟沙星的浓度。
60.所述线性关系为:y=0.01709x+1.01243,r2=0.997,x为诺氟沙星浓度,x为0.5~50μmol/l,y为第二荧光强度和第一荧光强度的比值。
61.测试环境的ph为2~6,可以但是不限于取值为2、3、4、5、6中的任意一个。
62.待测液和n,s-cds水溶液的反应温度为20~30℃,可以但是不限于取值为20℃、22℃、24℃、26℃、28℃、30℃中的任意一个。
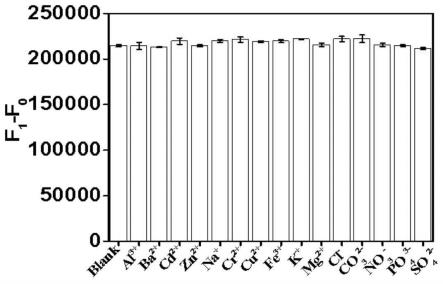
63.待测液和n,s-cds水溶液的反应时间为1~5min,可以但是不限于取值为1min、2min、3min、4min、5min中的任意一个。
64.以下通过具体的表征和试验来说明本发明的有益效果。
65.首先对不同柠檬酸铵和l-半胱氨酸的质量比、热处理温度和热处理时间制备得到的n,s-cds水溶液的使用效果进行了对比。其余制备参数为:每10ml水中加入1gl-半胱氨酸;采用10ml水溶解热处理产物后进行离心,离心处理为在100000rmp下离心5min;滤针的孔径为0.22μm;透析时间为24h;荧光检测时,n,s-cds水溶液的体积为250μl(下同),nor溶液体积为250μl,nor溶液的浓度为50μmol/l。
66.图1为n,s-cds水溶液的f
1-f0随柠檬酸铵和l-半胱氨酸的质量比的变化曲线。图2为n,s-cds水溶液的f
1-f0随热处理温度的变化曲线。图3为n,s-cds水溶液的f
1-f0随热处理时间的变化曲线。
67.从图1-3可以看出,n,s-cds水溶液的最佳制备工艺参数为:柠檬酸铵和l-半胱氨酸的质量比为2,热处理温度为180℃,热处理时间为60min。
68.图4为n,s-cds粉末的tem照片和hr-tem照片(插图)。图5为最佳制备工艺参数下所得n,s-cds的粒径分布图。
69.如图4所示,n,s-cds粉末以纳米球形颗粒高度分散,晶格间距为0.23nm,与石墨碳(sp2)的衍射面对应。
70.通过使用image j软件对图4中纳米球形颗粒进行粒径统计分析得到图5所示的纳米颗粒尺寸分布直方图。如图5所示,n,s-cds粉末的颗粒直径分布在1~5nm范围内,平均颗粒直径约为2.62nm。
71.图6为n,s-cds粉末的傅里叶变换红外光谱(ft-ir)。
72.如图6所示,在3432cm-1
和3180cm-1
处的两个宽峰分别对应于-oh和-nh,在1581cm-1
和1405cm-1
处的两个尖峰分别对应于-coo和-c=o,在1250cm-1
和615cm-1
处的两个特征峰分别对应于-c=s和-c-h。
73.图7为n,s-cds粉末的x射线光电子能谱(xps)全能谱图。
74.如图7所示,n,s-cds的xps全能谱图在532.87ev、400.1ev、285.81ev、231.1ev和163.81ev处具有特征峰,分别对应于o1s、n1s、c1s、s2s和s2p。
75.图8为n,s-cds粉末在c1s的xps光谱。图9为n,s-cds粉末在n1s的xps光谱。图10为n,s-cds粉末在o1s的xps光谱。图11为n,s-cds粉末在s2p的xps光谱。
76.如图8所示,c1s的xps光谱在284.8ev、286.1ev和288.1ev处的特征峰分别对应于c-c/c=c、c-n/c-o/c-s和c=o。如图9所示,n1s的xps光谱在398.8ev和400.9ev处的特征峰分别对应于c-n-c和h-n=c。如图10所示,o1s的xps光谱在531.2ev、532.1ev和533.1ev处的特征峰分别对应于c=o、c-o-h和c-o-c。如图11所示,s2p的xps光谱在163.2ev、164.2ev处的特征峰分别对应于s2p3/2的c-s-c和s2p1/2的c-s-c。
77.图12为n,s-cds水溶液以及n,s-cds+nor(n,s-cds水溶液和诺氟沙星溶液的混合物)的荧光寿命图。
78.如图12所示,用双指数函数拟合荧光衰减得到:n,s-cds-nor的荧光寿命为1.94ns,n,s-cds的荧光寿命为8.25ns。
79.在上述的表征中:
80.tem和hrtem采用fei talos f200x透射电子显微镜;ft-ir采用美国珀金莱默傅里叶变换红外光谱;xps采用德国热科k-alpha x射线光电子能谱;荧光寿命测试采用fls1000稳态/瞬态荧光光谱仪(英国爱丁堡)。
81.其次,对不同检测条件下n,s-cds水溶液的使用效果进行了对比。
82.图13为n,s-cds水溶液的f
1-f0随测试环境的ph的变化曲线。图14为n,s-cds水溶液的f
1-f0随反应温度的变化曲线。图15为n,s-cds水溶液的f
1-f0随反应时间的变化曲线。nor溶液体积为250μl,浓度为50μmol/l。在将n,s-cds水溶液与诺氟沙星溶液混合后用br缓冲液调节测试环境的ph。
83.从图13-15可以看出,n,s-cds水溶液的最佳使用条件参数为:测试环境的ph=4,n,s-cds水溶液与诺氟沙星溶液的反应时间为1min,反应温度为20℃。
84.在图1-3和图12-15中,f0为n,s-cds水溶液的荧光强度,f1为n,s-cds+抗生素(即n,s-cds水溶液和抗生素溶液的混合物)的荧光强度。
85.图16为n,s-cds水溶液检测不同抗生素的效果对比图。每种抗生素的用量为250μl,浓度均为50μmol/l,f0为n,s-cds水溶液的荧光强度,f1为n,s-cds+抗生素的荧光强度。
86.如图16所示,在含有cip(环丙沙星)、enr(恩诺沙星)、ofx(氧氟沙星)、smz(磺胺甲恶唑)或tc(四环素)的待测液中加入n,s-cds水溶液前后的荧光变化差值与在纯水中加入n,s-cds分散液前后的荧光变化差值几乎相同,说明n,s-cds能够特异性识别诺氟沙星。
87.图17为n,s-cds水溶液的抗干扰性能的测试结果。f0为n,s-cds水溶液的荧光强度,f1为n,s-cds+nor+干扰离子的荧光强度;干扰离子和nor共存时,干扰离子溶液和nor溶液体积为125μl,浓度均为50μmol/l;对照时采用125μl水替代干扰离子溶液。
88.如图17所示,诺氟沙星和干扰离子共存时,荧光变化程度(即f
1-f0)基本保持稳定,说明n,s-cds对诺氟沙星检测具有良好的抗干扰能力,可以应用于实际样品的检测中。
89.然后,对n,s-cds水溶液测试诺氟沙星的灵敏度、线性范围和实用性进行了测试。
90.图18为n,s-cds水溶液测试不同浓度的诺氟沙星溶液的荧光谱图。图19为n,s-cds水溶液的f1/f0与诺氟沙星浓度之间的线性拟合曲线。f0为n,s-cds水溶液的荧光强度,即第一荧光强度,f1为n,s-cds+nor的荧光强度,即第二荧光强度。
91.从图18可以看出,随着诺氟沙星浓度的增大,n,s-cds+nor的的荧光强度逐渐提升。从图19可以看出,f1/f0与诺氟沙星的浓度呈现良好的线性关系,当诺氟沙星浓度为0.5~50μmol/l时,回归方程为y=0.01709x+1.01243,r2=0.997,诺氟沙星的检出限为0.5μmol/l。
92.选取西南交通大学校园湖水以及实验室自来水作为实际样品,离心10min后用0.45μm的滤膜过滤,然后向水样中加入不同浓度的诺氟沙星(5μmol/l、20μmol/l、50μmol/l),采用n,s-cds和上述的回归方程测试水样中诺氟沙星的浓度。表1显示了n,s-cds对实际水样中诺氟沙星的检测结果。
93.表1
[0094][0095]
从表1可以看出,诺氟沙星的回收率在90%~104%范围内,且相对标准偏差(rsd)均小于2%,说明n,s-cds在实际水样中检测诺氟沙星的准确性较高,具有极大的实际应用价值。
[0096]
在上述的荧光测试中,在比色皿中,采用br缓冲液将待进行荧光检测的液体体积定容至1.5ml后再进行测试;测试环境的ph即指荧光测试时比色皿内液体的ph;待测液为单纯的抗生素时,f0获取时采用相同体积的水替代抗生素溶液。荧光测试时的激发光波长λex=350nm,激发和发射狭缝分别为3nm和1nm。
[0097]
以上对本发明的有关内容进行了说明。本领域普通技术人员在基于这些说明的情况下将能够实现本发明。基于本发明的上述内容,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1