一种检测植物源产品中氟吡草酮及其代谢物残留量的方法与流程
1.本技术涉及化学分析领域,具体涉及一种检测植物源产品中氟吡草酮及其代谢物残留量的方法。
背景技术:
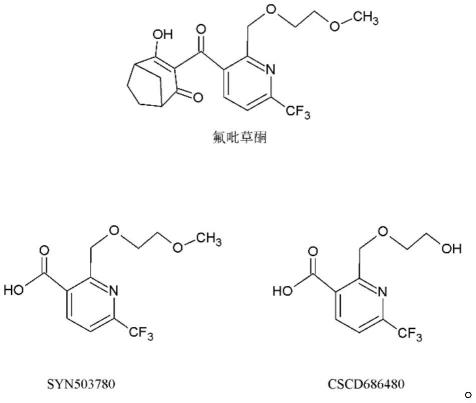
2.氟吡草酮(bicyclopyrone)是近年来先正达开发的新型4-羟基苯基丙酮酸双加氧酶(4-hppd)抑制剂类除草剂,主要用于玉米、甜菜和谷物等作物田。4-hppd是一种非血红素铁(ii)依赖的双加氧酶。氟吡草酮的作用是阻断4-hppd的功能,从而抑制类胡萝卜素的生物合成,使植物分生组织出现白化症状,最终致其死亡。氟吡草酮是一种广谱除草剂,具有非常优秀的选择性,主要防除玉米、甘蔗、谷物(如小麦、大麦)等作物田阔叶杂草和部分禾本科杂草,对三裂叶豚草和苍耳等大型种子阔叶杂草的防效较高,对草甘膦抗性杂草防效较好。氟吡草酮在植物和动物体内常代谢为2-(2-甲氧基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(syn503780)和2-(2-羟基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(cscd686480)。氟吡草酮的应用具有非常大的灵活性,从播种前到出苗后都能使用,另外它在不同的环境条件和不同的种植方式下也能够很好地发挥作用,氟吡草酮自2015年上市,现已在美国、加拿大、阿根廷、乌拉圭、澳大利亚等多个国家登记和上市,基于其优秀的特性而广泛应用,其中澳大利亚和美国制定了大麦、小麦、大田玉米、甘蔗等植物源性产品中的残留限量值,限量值低至0.02mg/kg,2022年美国环保署修订了氟吡草酮的残留限量,新增了在香蕉、洋葱、草莓、西瓜等12种产品中的残留限量,限量值低至0.01mg/kg,我国食品安全国家标准gb 2763-2021规定了大麦、小麦、玉米、鲜食玉米、麦胚和甘蔗中的残留限量,残留限量均以氟吡草酮及代谢物syn503780和cscd686480之和。
3.针对氟吡草酮的研究主要集中在除草活性、环境行为和安全性评价等方面,对于氟吡草酮的残留分析方法国内外相关报道较少。张强等采用高效液相色谱建立了44%氟吡草酮
·
精异丙甲草胺乳油高效液相色谱分析方法,对于氟吡草酮在植物源性产品中的分析方法尚未见报道。目前,现有相关技术还没有报道如何实现植物源产品中的氟吡草酮及其代谢物残留量进行定性筛查及定量分析。
技术实现要素:
4.为解决上述问题,本技术提供一种检测植物源产品中氟吡草酮及其代谢物残留量的方法。
5.本技术提供一种检测植物源产品中氟吡草酮及其代谢物残留量的方法,其包括:
6.从植物源产品中提取提取液;
7.对所述提取液进行除杂净化;
8.对除杂净化后的所述提取液进行质谱分析,得到植物源产品中氟吡草酮及其代谢物的残留量。
9.可选的,在本技术的一些实施例中,所述质谱分析包括:采用超高效液相色谱-四
极杆轨道肼高分辨质谱对所述提取液进行分析,其中,采用的色谱柱为硅胶基质色谱柱,采用流动相为含有体积百分浓度为0.2%-0.25%甲酸的水溶液,采用梯度洗脱分离。
10.可选的,在本技术的一些实施例中,所述硅胶基质色谱柱为waters acquity uplc hss t3型号的色谱柱,规格为2.1mm
×
100mm,粒径为1.8μm。
11.可选的,在本技术的一些实施例中,所述色谱柱的条件为流速:0.4ml/min-0.5ml/min;进样量:2μl-3μl。
12.可选的,在本技术的一些实施例中,所述超高效液相色谱-四极杆轨道肼高分辨质谱的工作条件:离子化方式为hesi;喷雾电压为3000v-4000v;毛细管的温度为300℃-400℃;离子传输管的温度为300℃-400℃;采集方式:在平行反应监测模式下进行,极性模式为正离子;鞘气:30arb-40arb;辅助气:5arb-10arb;质谱的扫描参数:二级扫描分辨率为17500dpi-35000dpi;二级质谱碰撞能为归一化碰撞能量,所述归一化碰撞能量为20%、40%或60%。
13.可选的,在本技术的一些实施例中,所述从植物源产品中提取提取液包括:
14.对植物源产品进行预处理,得到植物源试样;
15.将所述植物源试样、盐析剂和有机溶剂进行混合并进行提取,得到提取液。
16.可选的,在本技术的一些实施例中,所述有机溶剂为甲酸丙酮、甲酸乙酸乙酯、乙腈或甲酸乙腈中的任一种;和/或,所述盐析剂为氯化钠、无水硫酸钠或无水硫酸镁中的任一种或多种混合。
17.可选的,在本技术的一些实施例中,所述有机溶剂为体积百分浓度为0.4%-0.6%的甲酸乙腈。
18.可选的,在本技术的一些实施例中,所述植物源试样与所述盐析剂的质量比为(1-1.5):1;所述植物源试样与所述有机溶剂的质量体积比为1:(4-5)。
19.可选的,在本技术的一些实施例中,所述除杂净化采用分散固相萃取:将所述提取液、石墨化炭黑、十八烷基键合硅胶和无水硫酸镁混合,经离心处理后取上清液,过滤后得到除杂净化后的所述提取液。
20.本技术具有如下有益效果:
21.本技术,创新性的建立了对植物源性产品中氟吡草酮及其代谢物残留的检测方法,首次实现对氟吡草酮及其代谢物的定性筛查及同步定量;本技术的方法操作简单、灵敏度高及准确性好,适用于植物源产品中氟吡草酮及其代谢物的检测,能够为氟吡草酮及其代谢物在植物源性产品中的风险监控提供技术支撑。
附图说明
22.为了更清楚地说明本技术实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
23.图1为氟吡草酮在waters acquity uplc hsst3色谱柱上的二级质谱定量子离子(324.07730)提取离子色谱图;
24.图2为氟吡草酮在thermosyncronis c
18
色谱柱上的二级质谱定量子离子
(324.07730)提取离子色谱图;
25.图3为氟吡草酮在thermoaccucoreaq色谱柱上的二级质谱定量子离子(324.07730)提取离子色谱图;
26.图4为氟吡草酮(1ng/ml)的二级质谱定量子离子的提取离子色谱图;
27.图5为代谢物syn503780(1ng/ml)的二级质谱定量子离子提取离子流色谱图;
28.图6为代谢物cscd686480(1ng/ml)的二级质谱定量子离子提取离子流色谱图;
29.图7为在prm模式下获得的氟吡草酮(1ng/ml)的二级质谱图;
30.图8为在prm模式下获得的代谢物syn503780(1ng/ml)的二级质谱图;
31.图9为在prm模式下获得的代谢物cscd686480(1ng/ml)的二级质谱图;
32.图10为不同提取溶剂对氟吡草酮及其代谢物syn503780、cscd686480的提取回收率的影响示意图;
33.图11为不同提取溶剂的用量对氟吡草酮及其代谢物syn503780、cscd686480提取回收率的影响示意图;
34.图12为不同盐析剂对样品中氟吡草酮及其代谢物syn503780、cscd686480回收率的影响示意图;
35.图13为不同吸附剂的用量对氟吡草酮代谢物syn503780、cscd686480提取回收率的影响示意图;
36.图14为不同基质中氟吡草酮及其代谢物syn503780、cscd686480的基质效应示意图。
具体实施方式
37.下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本技术一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。
38.本技术的实施例提供一种检测植物源产品中氟吡草酮及其代谢物残留量的方法,涉及的植物源产品可以为但不限定小麦、大麦、麦胚、玉米、大葱、香蕉、甘蔗、洋葱、草莓、西瓜。氟吡草酮在植物体的代谢物为2-(2-甲氧基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(syn503780)和2-(2-羟基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(cscd686480),氟吡草酮及其代谢物(syn503780)和(cscd686480)的结构式如下:
[0039][0040]
检测方法包括:
[0041]
从植物源产品中提取提取液,该提取液中可能含有待测目标物氟吡草酮和/或氟吡草酮的代谢物。在一实施例中,采用溶剂法从植物源产品中提取提取液。
[0042]
对提取液进行除杂净化。在一实施例中,采用分散固相萃取净化去除提取液中的杂质。
[0043]
对除杂净化后的提取液进行质谱分析,得到植物源产品中氟吡草酮及其代谢物的残留量。在一些实施例中,质谱分析包括:采用超高效液相色谱-四极杆轨道肼高分辨质谱对提取液进行分析,其中,采用的色谱柱为硅胶基质色谱柱,采用流动相为含有体积百分浓度为0.2%-0.25%甲酸的水溶液,采用梯度洗脱分离。在其他一些实施例中,流动相可以为体积百分浓度为0.2%、0.21%、0.22%、0.23%、0.24%或0.25%甲酸的水溶液。经过试验表明,在流动相体系添加甲酸后对氟吡草酮及其代谢物syn503780和cscd686480的峰拖尾现象有明显改善,试验还证明了当水相中的甲酸的体积百分浓度达到0.2%时,氟吡草酮及其代谢物syn503780和cscd686480三种物质的峰形能达到较好的状态。
[0044]
超高效液相色谱-四极杆/静电场轨道阱高分辨质谱(ultrahigh performance liquid chromatography-quadrupole/orbitrap high resolution mass spectrometry,uplc-q-orbitrap hrms)将具有高选择性的四极杆与高分辨率、高灵敏度的轨道阱有机结合,能进行目标物与非目标物的快速筛查与结果确证,与低分辨三重四极杆质谱使用多反应监测模式(mrm)进行定量分析相比,uplc-q-orbitrap hrms无需对目标化合物子离子及相关质谱参数进行逐个优化,同时还弥补了传统三重四极杆质谱检测化合物数量有限、假阳性误判的不足,在食品检测行业中已得到广泛应用。已有研究中,使用高分辨质谱技术对目标物进行定量分析主要采用全扫描模式(full scan,fs),该模式与三重四极杆的多反应监测模式(mrm)相比,无法获得较佳的仪器方法灵敏度。在应用四极杆/静电场轨道阱高分辨质谱(q exactive)的平行反应监测(parallel reaction monitoring,prm)模式,仪器方法的灵敏度和fs模式相比得到了较大改善,可与三重四极杆的mrm模式相媲美。
[0045]
本实施例提供的检测方法的灵敏度满足国际国内相关法规的残留限量要求,可弥补在植物源性产品中氟吡草酮及其代谢物没有检测方法的空白;对评估和管理该化合物对人类健康和环境安全的影响,防止贸易输入风险具有重要作用。
[0046]
在本技术的一些实施例中,硅胶基质色谱柱为waters acquity uplc hss t3型号
的色谱柱,规格为2.1mm
×
100mm,粒径为1.8μm。waters acquity uplc hss t3色谱柱为高强度硅胶基质色谱柱,基于全多孔硅胶基质键合相,其保留性能优于普通c
18
柱,能够得到较好的峰形及响应,从而能够获得更好的峰形和更高的响应。
[0047]
在本技术的一些实施例中,色谱柱的条件为流速:0.4ml/min-0.5ml/min;进样量:2μl-3μl。在一优选地实施例中,色谱柱的流速为0.4ml/min,进样量为2μl。
[0048]
在本技术的一些实施例中,超高效液相色谱-四极杆轨道肼高分辨质谱的工作条件:离子化方式为hesi;喷雾电压为3000v-4000v;毛细管的温度为300℃-400℃;离子传输管的温度为300℃-400℃;采集方式:在平行反应监测模式下进行,极性模式为正离子;鞘气:30arb-40arb;辅助气:5arb-10arb;质谱的扫描参数:二级扫描分辨率为17500dpi-35000dpi;二级质谱碰撞能为归一化碰撞能量,归一化碰撞能量为20%、40%或60%。
[0049]
在一具体示例中,超高效液相色谱-四极杆轨道肼高分辨质谱的工作条件为:离子化方式为hesi;喷雾电压为3500v;毛细管的温度为350℃;离子传输管的温度为350℃;采集方式:在平行反应监测模式下进行,极性模式为正离子;鞘气:35arb;辅助气:10arb;质谱的扫描参数:二级扫描分辨率为17500dpi;二级质谱碰撞能为归一化碰撞能量,归一化碰撞能量为20%、40%或60%。
[0050]
在本技术的一些实施例中,从植物源产品中提取提取液包括:
[0051]
对植物源产品进行预处理,得到植物源试样。在一些实施例中,对植物源产品进行预处理是指:将植物源产品如稻谷、小麦、玉米(干)等粮谷类样品用粉碎机粉碎后使其全部可通过425μm的标准网筛。将蔬菜和水果样品用刀式研磨仪5000r/min粉碎并充分混匀。
[0052]
将植物源试样、盐析剂和有机溶剂进行混合并进行提取,得到提取液。在一具体示例中,称取5g(精确至0.01g)制备好的植物源试样于50ml聚丙烯离心管中,粮谷类样品预先加10ml水涡旋混匀,静置30min;加入5g氯化钠、25ml 0.4%甲酸乙腈溶液及1颗陶瓷均质子,涡旋振荡提取5min,5000r/min离心3min。吸取1.5ml上清液于2ml聚丙烯离心管中。
[0053]
在本技术的一些实施例中,有机溶剂为甲酸丙酮、甲酸乙酸乙酯、乙腈或甲酸乙腈中的任一种;和/或,盐析剂为氯化钠、无水硫酸钠或无水硫酸镁中的任一种或多种混合。
[0054]
在本技术的一些实施例中,有机溶剂为体积百分浓度为0.4%-0.6%的甲酸乙腈。采用0.4%-0.6%的甲酸乙腈,有利于提高氟吡草酮及其代谢物syn503780和代谢物cscd686480三种物质的回收率,从而有利于提高提取效率。在一优选实施例中,有机溶剂为体积百分浓度为0.4%的甲酸乙腈。
[0055]
在本技术的一些实施例中,植物源试样与盐析剂的质量比为(1-1.5):1。在其他一些实施例中,植物源试样与盐析剂的质量比还可以为1:1,1.2:1,1.3:1,1.4:1或1.5:1。植物源试样与有机溶剂的质量体积比为1:(4-5)。在其他一些实施例中,植物源试样与有机溶剂的质量体积比为1:4,1:4.5或1:5。
[0056]
在本技术的一些实施例中,除杂净化采用分散固相萃取:将提取液、石墨化炭黑、十八烷基键合硅胶和无水硫酸镁混合,经离心处理后取上清液,过滤后得到除杂净化后的提取液。植物源性样品中常用的quechers净化试剂有gcb、c
18
、psa和nh2等,其中psa和nh2的作用机理相似,均具有弱阴离子交换能力,通过氢键与化合物产生作用,可有效去除样品中的有机酸、极性色素、脂肪酸、糖类以及其他能形成氢键的成分;c
18
去除如挥发油、萜类、脂类等非极性化合物;gcb对样品中极性和非极性有机干扰物有极高的吸附力,对去除植物中
色素效果显著;无水硫酸镁可去除样液中的水分。实验考察了gcb、psa、c
18
和mgso4三种净化试剂对5μg/l氟吡草酮标准溶液进行吸附后的回收率。结果表明,经psa净化后氟吡草酮及其代谢物syn503780和代谢物cscd686480三种物质的回收率均低于80%,并随用量的增加回收率下降更多;gcb、c
18
、mgso4对三种物质的平均吸附回收率均在90%-110%之间。
[0057]
在另一些实施例中,采用quechers(quick、easy、cheap、effective、rugged、safe)方法,以甲酸乙腈溶液作为提取溶剂,经盐析后,提取液经分散固相萃取净化,基于超高效液相色谱-四极杆/静电场轨道阱高分辨质谱的prm模式,建立了植物源性产品中氟吡草酮及其代谢物残留的检测方法,同时建立了氟吡草酮及其代谢物的二级质谱数据库,实现对氟吡草酮及其代谢物的定性筛查及同步定量。该方法的灵敏度满足国际国内相关法规的残留限量要求,可弥补我国在植物源性产品中氟吡草酮及其代谢物没有检测方法的空白;对评估和管理该化合物对人类健康和环境安全的影响,防止贸易输入风险具有重要作用。
[0058]
为使本发明上述实施细节和操作能清楚地被本领域技术人员理解,以及本发明实施例的检测植物源产品中氟吡草酮及其代谢物残留量的方法的进步性能显著的体现,以下通过实验例来举例说明上述技术方案。
[0059]
实验例1
[0060]
试剂与仪器的选用:
[0061]
无水硫酸镁(mgso4)、无水硫酸钠(na2so4)、氯化钠(nacl)、正己烷、丙酮、乙酸乙酯均为分析纯,购于国药集团化学试剂有限公司;乙腈和甲醇均为色谱纯,购于美国tedia公司。石墨化炭黑(gcb,40-120μm)、乙二胺-n-丙基硅烷化硅胶(psa,40-60μm)和十八烷基键合硅胶(c
18
,40-60μm)购于上海安谱实验科技股份有限公司。微孔滤膜:0.22μm,有机相型;陶瓷均质子:2cm(长)
×
1cm(外径)。氟吡草酮(c
19h20
f3no5,100μg/ml,bepure公司)。氟吡草酮代谢物syn503780:2-(2-甲氧基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(c
11h12
f3no4,100μg/ml,bepure公司);氟吡草酮代谢物cscd686480:2-(2-羟基乙氧甲基)-6-(三氟甲基)吡啶-3-羧酸(c
10h10
f3no4,纯度97.0%,德国dr.ehrenstorfer公司)。
[0062]
q-exactive四极杆-静电场轨道阱高分辨质谱系统(美国thermo scientific);ultimate3000快速高效液相色谱系统(美国thermo scientific);milli-q纯水仪(美国millipore公司);粉碎机(上海嘉定粮油仪器有限公司);gm200刀式研磨仪(德国莱驰);涡旋振荡器(美国talboys公司);离心机(湖南湘仪实验室仪器开发有限公司)。
[0063]
标准溶液配制:
[0064]
称取适量cscd686480,用甲醇溶解配制成1000mg/l标准储备液。分别吸取适量的氟吡草酮、syn503780和cscd686480标准储备液,用甲醇稀释成氟吡草酮0.002mg/l,氟吡草酮代谢物syn503780和cscd686480 0.1mg/l的混合标准中间液;再用甲醇和空白样品提取液稀释混合标准中间液得到系列标准工作溶液和基质匹配标准工作溶液,其中,氟吡草酮浓度为0.002μg/l、0.004μg/l、0.02μg/l、0.04μg/l、0.2μg/l,氟吡草酮代谢物syn503780和cscd686480浓度为0.1μg/l、0.2μg/l、1μg/l、2μg/l、10μg/l,工作溶液现配现用。
[0065]
样品前处理:
[0066]
稻谷、小麦、玉米(干)等粮谷类样品用粉碎机粉碎后使其全部可通过425μm的标准网筛,蔬菜和水果样品用刀式研磨仪5000r/min粉碎并充分混匀。称取5g(精确至0.01g)制备好的试样于50ml聚丙烯离心管中,粮谷类样品预先加10ml水涡旋混匀,静置30min;加入
5g氯化钠、25ml 0.4%甲酸乙腈溶液及1颗陶瓷均质子,涡旋振荡提取5min,5000r/min离心3min。吸取1.5ml上清液于2ml聚丙烯离心管中,加入5mg gcb、5mg c
18
和50mg mgso4,涡旋混合1min,12000r/min离心3min,取上清液过0.22μm有机滤膜,待测定。
[0067]
仪器条件:
[0068]
色谱柱:waters acquity uplc hss t3液相色谱柱;流速:0.4ml/min;进样量:2μl;流动相a为0.2%甲酸水,流动相b为甲醇;梯度洗脱程序:0-2min,10%b;2-2.5min,10%-90%b;2.5-5min,90%b;5-5.1min,90%-10%b;5.1-7min,10%b。
[0069]
hesi离子化方式;喷雾电压为3500v;毛细管温度为350℃;离子传输管温度:350℃;采集方式:平行反应监测(prm)模式,正离子模式;鞘气(n2):35arb;辅助气(n2):10arb;二级扫描分辨率:17500;二级质谱碰撞能为归一化碰撞能(nce):20%、40%、60%。
[0070]
在上述质谱条件下,优化得到氟吡草酮及其代谢物的质谱参数见表1。
[0071][0072]
表1
[0073]
实验例2仪器条件的优化
[0074]
关于色谱柱的优化选择:
[0075]
本实验例对比了5μg/l的氟吡草酮及代谢物标准溶液在waters acquity uplc hsst3色谱柱(2.1mm
×
100mm
×
1.8μm)、thermosyncronis c
18
色谱柱(2.1mm
×
100mm
×
1.9μm)和thermoaccucoreaq色谱柱(2.1mm
×
150mm
×
2.6μm)上的峰宽和响应值,对于两种氟吡草酮代谢物syn503780和cscd686480,在三种色谱柱上峰形均良好;但对于氟吡草酮,在thermoaccucoreaq c
18
色谱柱上会出现较严重的峰展宽和拖尾,相对thermosyncronis c
18
色谱柱,在waters acquity uplc hss t3色谱柱上能获得更好的峰形和更高的响应,waters acquity uplchss t3色谱柱为高强度硅胶基质色谱柱,基于全多孔硅胶基质键合相,其保留性能优于普通c
18
柱,能够得到较好的峰形及响应。因此,waters acquity uplc hsst3色谱柱为较为优选地色谱柱。具体结果请参阅图1-图3所示,图1为氟吡草酮在waters acquity uplc hsst3色谱柱上的二级质谱定量子离子(324.07730)提取离子色谱图;图2为氟吡草酮在thermosyncronis c
18
色谱柱上的二级质谱定量子离子(324.07730)提取离子色谱图;图3为氟吡草酮在thermoaccucoreaq色谱柱上的二级质谱定量子离子(324.07730)提取离子色谱图。
[0076]
实验例3仪器条件的优化
[0077]
关于流动相的优化选择:
[0078]
实验例考察了不同流动相体系(乙腈-水、乙腈-0.2%甲酸水、甲醇-水、甲醇-0.1%甲酸水,甲醇-0.2%甲酸水和甲醇-5mmol/l乙酸铵水(含0.1%甲酸))对色谱峰形和信号响应的影响。结果表明,不含甲酸的流动相体系(乙腈-水和甲醇-水)中氟吡草酮及其代谢物syn503780和cscd686480的峰型均出现前伸和拖尾现象,流动相体系添加甲酸后峰拖尾现象得到明显改善,当水相中的甲酸含量达到0.2%时三种物质的峰形能达到较好的
状态,水相中添加乙酸铵对三种物质的响应和峰形无明显影响。实验结果表明乙腈-0.2%甲酸水和甲醇-0.2%甲酸水均可作为流动相,由于甲醇作流动相时,系统压力较小,所以最终选择甲醇-0.2%甲酸水为流动相。
[0079]
实验例4仪器条件的优化
[0080]
关于质谱条件的优化选择:
[0081]
利用uplc-q-orbitrap hrms的prm模式对氟吡草酮及其代谢物混合标准溶液进行检测,仅需提供各化合物的分子离子峰[m+h]
+
理论精确质量数,碰撞能量使用归一化碰撞能量(nce)设置为20%,40%,60%即可。prm采集模式首先利用四极杆质量分析器的选择检测能力,在一级质谱中选择性地检测目标化合物的母离子信息即分子离子峰[m+h]
+
,随后在碰撞池中对母离子进行碎裂;最后利用高质量精度的高分辨质量分析器在二级质谱中检测所选择母离子的所有碎片信息。prm技术与mrm技术很相似,不同的是二级质谱检测模式的不同,prm技术是在母离子碎裂后通过高分辨质量分析器对全部子离子进行检测,相对于mrm技术,通过监测子离子的精确质量数可进一步降低背景噪音,受到的离子噪音干扰远小于mrm技术,从而提高了仪器的灵敏度和准确性。同时prm技术在使用方面也更加简便,不需要寻找准确的母离子-子离子和预先对相关参数进行优化。使用prm模式对氟吡草酮及其代谢物syn503780、cscd686480进行检测,其结果请参照图4-图9所示。图4为氟吡草酮(1ng/ml)的二级质谱定量子离子的提取离子色谱图;图5为代谢物syn503780(1ng/ml)的二级质谱定量子离子提取离子流色谱图;图6为代谢物cscd686480(1ng/ml)的二级质谱定量子离子提取离子流色谱图;图7为在prm模式下获得的氟吡草酮(1ng/ml)的二级质谱图;图8为在prm模式下获得的代谢物syn503780(1ng/ml)的二级质谱图;图9为在prm模式下获得的代谢物cscd686480(1ng/ml)的二级质谱图。
[0082]
实验例5前处理方法的优化
[0083]
关于提取溶剂的优化选择:
[0084]
对于植物源性试样,可以使用丙酮、乙酸乙酯、甲醇、乙腈作为提取溶剂。因甲醇提取液经盐析也无法与水相分离而排除甲醇为提取溶剂。实验选择0.4%甲酸丙酮、0.4%甲酸乙酸乙酯、乙腈、0.1%甲酸乙腈、0.2%甲酸乙腈和0.4%甲酸乙腈为提取溶剂,以氟吡草酮及其代谢物syn503780、cscd686480的平均回收率(加标量5μg/kg)考察不同溶剂的提取效果。对于乙腈和甲酸乙腈溶液,将添加5μg/kg氟吡草酮及其代谢物syn503780、cscd686480的小麦样品按照实验例1样品前处理方法处理后上机测定;每个实验平行测定3次,采用溶剂标准曲线进行校准定量;对于0.4%甲酸丙酮、0.4%甲酸乙酸乙酯,按照实验例1样品前处理方法处理后,分别取1ml提取液40℃氮吹后用乙腈复溶后上机。由图10可知,以0.4%甲酸丙酮为提取溶剂时,代谢物cscd686480回收率低;以0.4%甲酸乙酸乙酯为提取溶剂时,代谢物syn545910和代谢物cscd686480回收率较低;当乙腈为提取溶剂时,随着甲酸的含量增加,氟吡草酮及其代谢物syn503780、cscd686480的回收率也随之增加,并在0.4%甲酸乙腈提取时三种物质的回收率较好,提取效率较高。由此说明提取溶剂的ph值对提取效率的影响较大,研究得出选择0.4%甲酸乙腈作为提取溶剂,有利于提高提取效率。
[0085]
实验例6前处理方法的优化
[0086]
关于提取溶剂乙腈用量的优化:
[0087]
以小麦为实验样品,选择10ml、25ml、40ml和50ml进行实验优化乙腈用量,以最终
回收率作为考察依据。请参照图11,结果表明,提取溶剂为10ml时,由于基质效应的影响,代谢物的回收率相对偏低,乙腈用量为25ml时,回收率可达到分析测定要求,当用量继续增大,回收率变化不明显,同时会导致方法检出限升高。由此说明,最优选地提取溶剂乙腈用量为25ml,操作较为便捷,降低了耗材成本。
[0088]
实验例7前处理方法的优化
[0089]
关于盐析剂的优化选择:
[0090]
盐析剂的作用主要是去除样品中多余的水分,使农药组分在有机相中的溶解度增加,有利于提高提取效率。本实验测试了3种常用盐析剂,即氯化钠、无水硫酸钠、以及无水硫酸镁+氯化钠(3+1)混合盐析剂对样品中3种农药组分提取效果的影响,结果如图12所示。由图可知,盐析剂采用无水硫酸钠和无水硫酸镁+氯化钠(3+1)时,三种组分的提取回收率均低于以氯化钠为盐析剂时的提取回收率,由此说明,盐析剂为氯化钠时,盐析效果,有利于提高提取液的回收率,同时氯化钠价格低,降低成本。
[0091]
实验例8前处理方法的优化
[0092]
关于净化试剂的优化选择:
[0093]
植物源性样品中常用的quechers净化试剂有gcb、c
18
、psa和nh2等,其中psa和nh2的作用机理相似,均具有弱阴离子交换能力,通过氢键与化合物产生作用,可有效去除样品中的有机酸、极性色素、脂肪酸、糖类以及其他能形成氢键的成分;c
18
去除如挥发油、萜类、脂类等非极性化合物;gcb对样品中极性和非极性有机干扰物有极高的吸附力,对去除植物中色素效果显著;无水硫酸镁可去除样液中的水分。实验考察了gcb、psa、c
18
和mgso
4 3种净化试剂对5μg/l氟吡草酮标准溶液进行吸附后的回收率。结果表明,psa净化后三种物质的回收率均低于80%,并随用量的增加回收率下降更多;gcb、c
18
、mgso4对三种物质的平均吸附回收率均在90-110%之间,由此说明,选用gcb、c
18
、mgso4作为净化试剂,有利于提高提取液的回收率。
[0094]
实验例9前处理方法的优化
[0095]
关于净化试剂组合用量的确定:
[0096]
植物源性样品使用分散固相萃取进行净化时通常使用两种或多种净化试剂,以较好地除去样品中的脂类和糖类等干扰杂质。实验考察了3组净化试剂(ⅰ:0mg gcb+0mg c
18
+50mg mgso4,ⅱ:5mg gcb+5mg c
18
+50mg mgso4,ⅲ:10mg gcb+10mg c
18
+50mg mgso4,ⅳ:20mg gcb+20mg c
18
+50mg mgso4)对小麦空白样品5μg/kg加标提取液的净化效果,如图13所示,结果表明,3组净化试剂的回收率均在90-110%之间,均可满足实验要求;从实际净化效果看,随着gcb和c
18
用量的增加,氟吡草酮及其代谢物syn503780、cscd686480的回收率变化较小,组合ⅰ就可以满足绝大部分样品对色素的清除。由此说明,选择净化试剂组合为5mg gcb+5mg c
18
+50mg mgso4,有利于提高净化效果,同时降低综合成本。
[0097]
实验例10基质效应的影响
[0098]
uplc-q-orbitrap hrms具有更强的抗干扰能力,但基质效应仍然存在,且植物源性样品众多,基质复杂,因此,需要对基质效应进行评价,基质效应(matrix effects,me)在高分辨质谱系统主要由样品中目标物以外的组分,与目标物共洗脱出的样品基质对目标物的离子化过程产生影响,造成离子化抑制或增强。实验按照实验例1样品前处理方法制备空白基质溶液,按照实验例标准溶液配制方法配制标准工作溶液和基质标准工作溶液上机测
定。按照下式计算基质效应:me=[(基质匹配校准曲线斜率/纯溶剂标准曲线斜率)-1]
×
100%,如图14所示,结果表明稻谷、小麦、玉米、甘蔗、香蕉、大葱、西蓝花和西瓜中氟吡草酮及其代谢物的基质效应在1%-20%之间,基质效应均小于20%,氟吡草酮及其代谢物在这6种基质中基质效应均表现为弱基质效应,无需补偿基质效应。
[0099]
实验例11方法学评价
[0100]
关于线性关系与定量下限:
[0101]
按照实验例1标准溶液配制方法配制氟吡草酮及其代谢物系列标准工作溶液和基质匹配标准工作溶液,按照实验例1仪器条件进样检测,以质量浓度(x,μg/l)为横坐标,以其峰面积为纵坐标(y)绘制标准工作曲线。在空白样品溶液中添加适量的标准溶液后上机测定,以10倍信噪比(s/n=10)确定定量下限(loq),氟吡草酮及其代谢物的线性回归方程、相关系数(r)、线性范围及定量限见表2,表2为氟吡草酮及其代谢物的回归方程、相关系数、线性范围和定量限。
[0102][0103][0104]
表2
[0105]
表2中,y:peak area of quantitative ion;x:mass concentration,μg/l。
[0106]
实验例12回收率与相对标准偏差
[0107]
分别对小麦、玉米、大葱、香蕉和甘蔗空白样品进行低、中、高3个水平的加标回收实验,每个加标浓度测定6次平行,回收率和相对标准偏差(rsd,n=6)见表3。氟吡草酮及其代谢物syn503780、cscd686480在3个加标水平下的平均回收率为84.6%-118.5%,rsd为2.6%-7.4%,由此表明,本技术提供的检测植物源产品中氟吡草酮及其代谢物残留量的方法,具有较好的准确度和精密度。
[0108][0109]
表3
[0110]
实际样品检测
[0111]
采用本方法对市场采购样品小麦、玉米、大葱、香蕉、甘蔗各5个进行检测,采用精
确质量数和保留时间对样品中的氟吡草酮及其代谢物定性筛查,并结合二级特征碎片离子确证,在一个进境玉米种检出氟吡草酮残留。
[0112]
本技术上述实施例建立了超高效液相色谱-四极杆静电轨道阱高分辨质谱法测定植物源性产品中氟吡草酮及其代谢物残留量的分析检测方法,在优化实验条件下,氟吡草酮在0.002-0.2μg/l、代谢物syn503780和代谢物cscd686480在0.1-10μg/l范围内线性关系良好,相关系数均大于0.995。空白样品在低、中、高3个水平的加标的平均回收率为84.6%-118.5%,rsd为2.6%-7.4%,氟吡草酮定量限为0.01μg/kg,代谢物syn503780和cscd686480定量限为0.5μg/kg。该方法灵敏度高、操作简单、快速、准确,能满足植物源性产品中氟吡草酮及其代谢物残留量的检测需求。
[0113]
通过对仪器条件、提取溶剂和净化方式等条件进行优化,确定最合适的前处理方法和仪器条件。对样品的基质效应进行考察发现,氟吡草酮及其代谢物在8种样品中的基质效应均小于20%,为弱基质效应,可无需采取补偿措施。在优化实验条件下,氟吡草酮在0.002-0.2μg/l、代谢物syn503780和代谢物cscd686480在0.1-10μg/l范围内线性关系良好,相关系数均大于0.995。空白样品在低、中、高3个加标水平下的平均回收率为84.6%-118.5%,相对标准偏差(n=6)为2.6%-7.4%,氟吡草酮定量限为0.01μg/kg,代谢物syn503780和代谢物cscd686480定量限为0.5μg/kg。该方法灵敏度高、操作简单、快速、准确,能满足植物源性产品中氟吡草酮及其代谢物残留量的检测需求。采用该方法对25个样品进行测定,在一个进境玉米样品中检出氟吡草酮残留。该方法操作简单、灵敏度高、准确性好,适用于植物源产品中氟吡草酮及其代谢物的检测,可为氟吡草酮及其代谢物在植物源性产品中的风险监控提供技术支撑。
[0114]
以上对本技术所提供的检测植物源产品中氟吡草酮及其代谢物残留量的方法进行了详细介绍,本文中应用了具体个例对本技术的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本技术的方法及其核心思想;同时,对于本领域的技术人员,依据本技术的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本技术的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1