测序结果中序列数据错误的检测和校正方法与流程
本发明涉及测序反应结果中序列数据错误的检测和校正方法,属于基因测序领域。
背景技术:
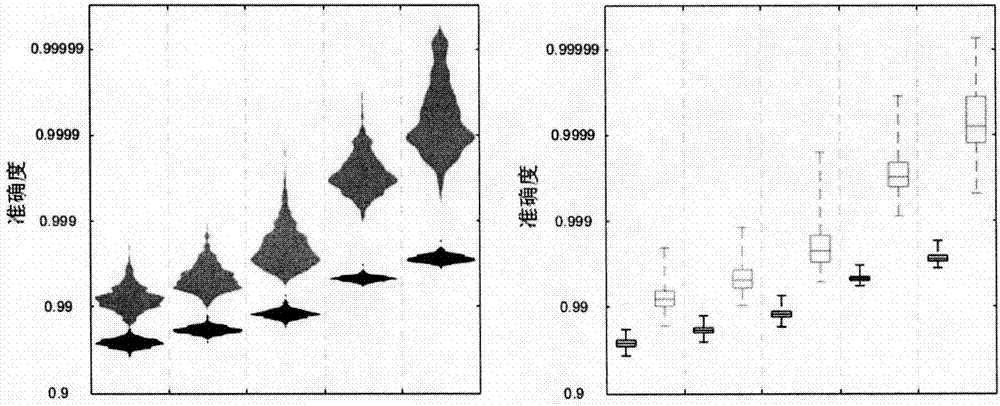
:近年来,随着人们对于基因的认识越来越深入,基因测序对于医学以及生物学带来了巨大的变革。传统的测序方法主要包括SangerDNA测序法、限制性酶切长度多态性、单链构象多态性和基于基因芯片的寡核苷酸探针杂交法等。测序过程中,由于种种的原因,例如CD采光不准、流体异动、环境光、杂DNA、信号校正系统误差、测序反应液不纯等原因,测序结果出现错误是不可避免的。DNA作为遗传物质,储存了生物的遗传信息,该特性亦使得DNA被用作一般信息的存储介质。在利用DNA存储信息时,需要将信息编码成DNA序列,然后利用基因测序的方法读取该信息。为了避免编码和/或读取中的错误,通常会在编码过程中引入冗余信息,并利用该冗余信息在读取中进行信号校正。例如乔治·彻奇等人使用里德-所罗门码来将信息编码成DNA序列,并使用Illumina测序平台来读取DNA序列上的信息。DNA编码-读取技术还被应用于组合化学等领域中。在以往的DNA编码技术中,每个碱基的类型通常与其他位置上的碱基无关(无记忆型编码),或者仅与其邻近范围内的碱基有关。本发明提出了一种有记忆的、分布式、正交DNA编码方式,其每个碱基的类型与它前面位置上所有碱基均有关,并利用多组正交编码之间的综合对比来进行解码,有效提高编码-读取过程的准确率。技术实现要素:本专利涉及测序反应结果中序列数据错误的检测和校正方法。本发明公开一种测序结果中编码错误的检测和校正方法,其特征在于,对同一核酸序列进行测序,得到三条或者三条以上相互正交的核酸简并序列数据,三条或者三条以上正交的核酸简并序列对比,可以检测到序列中的错误;进一步在对比出现错误的位置,通过修改至少一个序列,可以获得校正的序列。本发明公开一种测序结果中编码错误的检测和校正方法,其特征在于,对同一核酸序列进行测序,得到三条或者三条以上使用M、K、R、Y、W、S、B、D、H、V字母表示的简并序列,三条或者三条以上简并的序列对比,可以检测序列错误;进一步在对比出现错误的位置,通过修改至少一个序列,可以获得校正的序列。本方法适应常规的测序方法,只要测序底物设计合理,通过多轮测序,获得三条或者三条(轮)以上的编码结果,则可以利用其中信息的冗余,检测和校正错误的编码。本发明公开一种利用基因编码的记忆性检测和校正编码错误的方法,对同一核酸序列进行测序,得到两条或者两条以上的使用M、K、R、Y、W、S、B、D、H、V字母表示的简并序列,获得一个以A、G、T、C为编码的核酸序列信息,或者可以获得以A、G、U、C为编码的核酸序列信息;利用测序反应中由不同核苷酸碱基上连接的不同官能团导致的光或电信号作为冗余信息,可以检测序列错误;进一步在对比出现错误的位置,通过修改至少一个序列,可以获得校正的序列。本发明公开一种利用基因编码的记忆性检测和校正编码错误的方法,对同一核酸序列进行测序,获得三条或者三条以上相互正交的核酸简并序列数据;将简并序列综合对比,利用核酸序列的记忆性检测序列错误;进一步在对比出现错误的位置,通过修改至少一个序列,可以获得校正的序列;所述简并序列中,每个序列信号表示了部分基因序列信息,并且从其中一个简并序列上的信号,并不能推定另外一个简并序列上同一位置的信号。将待测核酸片段固定,通入反应液进行测序反应,每轮测序得到一条简并的核酸序列;至少经过N轮测序,获得N条简并的核酸序列;N条简并的序列综合对比,可以检测序列出现错误的位置;进一步在对比出现错误的位置,通过修改至少一个序列,可以获得校正的序列;其中,所述的反应液中,含有两种或者两种以上不同碱基的核苷酸底物分子;所述N为大于等于2的正整数。根据优选的技术方案,通过N-1条简并的核酸序列对比,可以获得一个以A、G、T、C为编码的核酸序列信息,或者可以获得以A、G、U、C为编码的核酸序列信息;利用N条简并的序列综合对比,可以检测序列出现错误的位置,所述N为大于等于3的正整数。根据优选的技术方案,通过N条简并的核酸序列对比,可以获得一个以A、G、T、C为编码的核酸序列信息,或者可以获得以A、G、U、C为编码的核酸序列信息;利用两种或两种以上的连接到碱基的官能团所提供的发光信息可以检测序列出现的错误的位置,所述N为大于等于2的正整数。或者利用测序反应中碱基本身信息变化或者反应过程中释放的磷酸、氢离子等分子的信息,可以作为冗余信息,用于校正。本发明公开一种测序结果中编码错误的检测和校正方法,其特征在于,将待测核酸片段固定,通入反应液进行测序反应;测序所用的核苷酸底物分子反应液根据碱基不同分成三组,每组反应液包含两个不同的反应液,每个反应液中含有两种不同碱基的核苷酸底物分子,两个反应液中核苷酸上的碱基种类没有交集;每轮测序使用一组反应液,每组反应液中的两个反应液循环进入;使用三组反应液进行三轮测序,得到三条简并的序列;三条简并的序列综合对比,可以检测序列出现错误的位置;进一步,在对比出现错误的位置,通过修改至少一个序列,获得校正的序列。根据本发明优选的替代技术方案,可以将所述的包含两种不同碱基的反应液简单的根据碱基的不同分成两个反应液;其它部分则可以做出相应的调整。根据本发明优选的实施方案,所述反应液为多个反应液,每次测序使用一种反应液;每轮测序使用一种或者一种以上的反应液;其中至少一种反应液中含有两种或者两种以上不同碱基的核苷酸底物分子;不同轮测序使用的反应液中含有不同的核苷酸底物分子组合。根据优选的技术方案,本发明所述测序指的是,利用5’端多磷酸修饰有荧光切换性质的荧光团的核苷酸底物分子进行测序;所述的荧光切换性质指的是测序后荧光信号相比测序反应前有明显改变;首先,将待测的核苷酸序列片段固定,然后通入含有核苷酸底物分子的反应液;使用酶将核苷酸底物上面的荧光团释放,从而导致荧光切换。根据本发明的方法,所述测序后荧光信号相比测序反应前有明显改变,指的是每一步的测序反应后,荧光信号相比于测序反应前有明显增强或者有明显减弱或者发射光频率范围有明显改变。根据本发明的技术方案,所述的序列错误指的是插入或缺失类型的错误。根据本发明的技术方案,序列数据错误指的是,当至少两条核酸序列信息在同一位置不表示一个共同的碱基的时候,认为出现错误。根据本发明的技术方案,校正序列错误,指的是将至少一个序列数据的错误进行修改,使得在之后的至少一个位置上的序列是正确的。所述序列是正确的指的是任意两轮序列再同一位置确定的核酸序列信息,同另外一轮序列获得的核酸序列信息不矛盾;或者说,任意两轮序列数据在同一位置表示的核酸序列信息,同连接到碱基的官能团所提供的发光信息或者其它测序过程中的信息不矛盾。根据本发明的技术方案,校正序列错误,也可以指的是将至少一个序列的错误进行修改,使得在之后的至少一个位置上的序列综合对比可以获得共同的碱基。根据本发明的技术方案,所述的修改至少一个序列指的是,将该代表核酸序列信息的序列在出错的位置,延长或者缩短序列,获得校正的序列。其中所述的延长或者缩短也可以指的是同一个信号的增大或者缩小;该位置的编码结果缩短或者延长的时候,其表示的基因序列的信息并没有变化;其结果是同一个编码,例如简并编码M信号强度为2,即MM;可以将其延长为3,即MMM。根据本发明的技术方案,所述核酸序列的记忆性指的是,测序结果中,某一位置的核酸序列信息,不仅和它所对应的待测核酸上的序列有关系,还和它前面的序列信息有关系。根据本发明另一个优选的技术方案,所述的延长或者缩短某个测序信号中,延长某个测序信号指的是,将该位置表示的基因序列延长,同时利用其它两轮的测序信号获得校正的核酸序列;缩短某个测序信号指的是,将该位置表示的基因序列缩短或者删除一定的长度,同时利用其它两轮的测序信号获得校正的核酸序列。根据本发明另一个优选的技术方案,根据碱基不同将反应液分为三组,其中所述的碱基指的是A、G、C、T四种碱基或者A、G、C、U四种碱基;其中碱基可以是甲基化、羟甲基化、醛基化和羧基化的碱基,也可以是非甲基化、非羟甲基化、非醛基化和非羧基化的碱基。根据本发明另一个优选的实施方式,可以将包含两种不同碱基的核苷酸底物反应液根据碱基的不同分成两个反应液根据本发明另一个优选的实施方式,可以用荧光标记核苷酸底物分子;在核苷酸底物分子的碱基上修饰荧光团或者修饰通过化学反应可以发生荧光变化的官能团;可以用一种所述的荧光团或官能团修饰不同碱基的核苷酸底物分子,也可以用多种不同的荧光团或官能团修饰不同碱基的核苷酸底物分子。根据本发明优选的技术方案,每轮测序获得一组简并的基因序列信息,所述的简并的基因序列信息,指的是,包含了可能的基因序列信息。例如:当反应液中含有A、G碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基C和/或T的基因序列信息;当反应液中含有A、T碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基C和/或G的基因序列信息;当反应液中含有A、C碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基G和/或T的基因序列信息;当反应液中含有C、G碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基A和/或T的基因序列信息;当反应液中含有C、T碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基A和/或G的基因序列信息;当反应液中含有T、G碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基C和/或A的基因序列信息。根据本发明优选的技术方案,当三轮测序的信号综合对比的时候,其中一轮测序的信号所表示的基因序列信息是一个偏大错误的测序信号,则可以缩短该测序信号表示的基因序列信息,使得后面至少1个测序信号的对比结果是正确的。根据本发明优选的技术方案,当三轮测序的序列综合对比的时候,其中一轮测序的序列信息所表示的基因序列信息是一个偏小错误的测序信号,则可以在该位置表示的基因序列信息添加空位,延长;使得后面至少1个测序信号的对比结果是正确的。比如获得M值的信号强度为2,即MM;可以将其延长为3,即MMM。本发明描述的是基因测序编码结果的错误检测和校正的方法。特别是每个反应液中包含了两种或者两种以上的碱基的核苷酸底物分子的测序方法。本发明适合于SBS(边合成边测序)的方法。本专利涉及的名词为本领域的常规含义,为了更清楚的表达,现特将所述含义做出一般性的解释。简并的基因序列信息,指的是,包含了可能的基因序列信息。例如,当反应液中含有A、G碱基的核苷酸底物分子的时候,测序获得的简并的基因序列信息中包含了待测的核苷酸序列上的碱基C和/或T的基因序列信息。假设测序反应得到的强度信息是3,其代表的含义是待测基因可能含有3个C和/或T的,例如3个C,或者3个T,或者1个C两个T,或者1个T两个C,其并不能区分C和T的前后位置。简并的基因序列信息或者说简并的编码属于本领域的常用词语。本发明所描述的方法虽然可以检测并校正测序中的错误,但并不能完全消除测序错误。有极低的概率,使得被修改的信号并不是真正发生错误的信号。可以通过以下方法进一步提高测序的最终准确率:如果将MK、RY、WS这三次信号中被修改的信号集中在一起,其中连续N次之内有两次信号均被修改,则认为解码中很可能发生了错误,并将该序列丢弃。此处N为大于或等于2的正整数。N越大,则丢弃序列的比例越高,最终解码准确率也越高。本发明优选的N值为3。本发明中,按照IUPAC符号命名规则(Nucleicacidnotation),使用下面表1的字母表示简并碱基,例如字母M表示A和/或T。字母所代表的碱基MACKGTRAGYCTWATSCGBCGTDAGTHACTVACG表1共聚物(copolymer):由两种不同脱氧核苷酸组成的DNA区域,如AAC、GGTG。本发明中所述的序列数据错误的检测和校正方法,指的是,可以检测序列错误的位置;也可以校正序列错误。实际测序的过程中,首先通过循环的测序反应,获得光学或者其它信号的相对强度值;该强度值可以用一定的形式表示,例如M,表示该位置碱基(可以多种碱基)的位置和数量等信息,可以表示简并的基因编码结果。足够信息量的相对强度值进行解码可以获得待测基因的基因序列信息。本发明中,所述呈递同时也是加入反应液的意思。本发明中,三轮或者多轮表示的时候,根据其环境的不同,可以替代为多轮或者两轮。按次计的测序信号:每一次测序反应的信号的强度信息。在理想情况下其表示的信息和对应共聚物长度相同。按位计的测序信号:依次读取按次计的测序信号,若该信号强度为n,且其反应液中加入的核苷酸为X,则写上n个X。例如图1中的按次计的测序信号转换为按位计的测序信号为MMMKKKKKMKKKMMK,或者写为(A/C,A/C,A/C,G/T,G/T,G/T,G/T,G/T,A/C,G/T,G/T,G/T,A/C,A/C,G/T)。例如凡奇数次则使用含有dA4P和dC4P(含有4个磷酸基团、末端磷酸标记有荧光基团的核苷酸)的测序反应液,凡偶数次则使用含有dG4P和dT4P的测序反应液。经过若干次反应后获得一组荧光信号值(表2)。亦可采用其他荧光标记核苷酸的组合来获得与靶DNA序列相关的荧光信号值。可能的组合实例例如:1.M/K式:凡奇数次呈递dA4P和dC4P,凡偶数次呈递dG4P和dT4P;或者二者反过来;2.R/Y式:凡奇数次呈递dA4P和dG4P,凡偶数次呈递dC4P和dT4P;或者二者反过来;3.W/S式:凡奇数次呈递dA4P和dT4P,凡偶数次呈递dC4P和dG4P;或者二者反过来。表2将三种不同核苷酸组合下获得的测序数据表示为按位计的测序信号,联立在一起。对每一位置,求三条按位计的测序信号在该位置上所表示核苷酸种类的交集,即可获得靶DNA的序列。此即信号解码的基本原理。例如,若M/K、R/Y、W/S三种组合得到的按次计的测序信号分别为(3,5,1,3,2,1)、(2,4,3,2,1,3)、(2,1,3,2,3,3,1),则可推断出序列为AACTTTGGATTGCCT。三轮测序反应的结果综合对比,指的是,将三轮测序结果的化学发光或者其他形式的强度信号转化为基因序列信息以后,在同一碱基位置的三轮测序的结果对比,如果三轮测序获得的结果表示是一致的,则认为该位置测序的结果是正确的;如果三轮测序获得的结果表示的基因序列信息不一致,则认为该碱基位置测序的结果是错误的。若由于CCD采光不准、流体异动、环境光、杂DNA、信号校正系统误差、测序反应液不纯等原因,某一次的按次计的测序信号偏大或偏小,则会造成按位计的测序信号在相应位置及后续某些位置上所表示核苷酸种类的交集为空,无法解出核苷酸种类。显然,按次计的测序信号所发生的错误会造成按位计的测序信号从发生错误的位置起发生整体的偏移,因此按位计的测序信号是一种有记忆的信号。利用按位计的测序信号有记忆的特点,可以校正测序信号中的错误。本发明提供了一种测序结果中序列数据错误的检测和校正的方法。测序反应液中包含至少两种不同碱基的核苷酸底物分子;获得简并的基因编码信息。两个或者两个以上简并的编码信息相互对比,本领域技术人员可以判断该位置的编码是否出现相互矛盾的情况。相比于同一个待测底物,用不同引物或者直接测多轮的方法,其更加简洁,并且一次实验设计即可完成。本发明的方法相比于同一待测基因,直接测序多轮的情况,是完全不同的方法。本发明提供的方法,如果只有两个相互正交的简并基因编码结果,则不存在校正的基础(加入颜色等冗余信息的情况除外)。本发明首次提出了这种在基因测序中,三个或者三个以上相互正交的简并编码结果的错误检测以及校验。本发明提供一种测序结果中序列数据错误的校正方法。特别是利用5’端多磷酸修饰有荧光切换性质的荧光团的核苷酸底物分子进行测序,该方法也被称为荧光切换测序法。当荧光切换测序方法与2+2测序方法联合使用的时候,其测序方法本身带来了很多的优势,例如读长更长,测序准确率达到99.99%,读长可以达到300bp;这是单纯的2+2测序或者荧光切换测序方法所不具备的;同时,还具备其它的优势,例如允许更高的通量,反应简单,错误率低,不需要实时采集信息等。相同的,荧光切换的其它多核苷酸底物分子测序也具备部分相同的性质。例如,荧光切换测序方法和2+2测序方法,当三轮测序的时候,提供了除颜色信息(发光信息或者其它可检测信息)以外的冗余信息可供校正;使得其可以在准确率不变的情况下延长了有效读长;这种校正的结果依赖于测序方法的准确度,在测序仪器准确率固定的情况下,大大提高了有效读长内的总体准确率;例如,400bp的核酸片段的测序正确率为97.36%,经过错误校正以后的正确率可以达到99.17%。因此,应用了该错误检测与校正方法的测序仪器,还可以相应的进一步延长有效读长。利用本发明提供的的方法进行校正,表现出明显的规律:任何对于测序方法的可以降低错误率的微小改进,都可以更加明显的降低校正以后的编码数据的错误率。附图说明图1,序列数据错误校正的图表表示方法。图2,用小提琴图和箱型图表示1-5组数据的数据分布规律。其中黑色表示编码准确率,灰色表示解码后准确率。从左到右依次代表1-5组数据。图3,频率分布直方图,统计了5000条序列数据中每条序列在解码中被修改的信号的数目。图4,编码中发生错误的信号的编号和解码中被错误地修改的信号的编号之间的相关关系,横坐标表示编码中发生错误的信号的编号,纵坐标表示解码中被错误地修改的信号的编号之间的相关关系,颜色的灰度表示该点被计数的次数占所有序列中的比例。具体实施方式为了进一步说明本发明的核心内容,现将本发明用下面的例子作为说明。实施例是为了进一步解释
发明内容部分,并不对于本发明造成限制。需要指出的是,本发明中涉及的所有具体序列或者测序步骤,均为本领域的普通技术知识。实施例1将待测单链DNA分子固定在固相表面上。固定的方法可以是化学交联、分子吸附等,可以把DNA的3’端或者5’端固定。该待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5′-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。其中,核苷酸底物分子的碱基上连接了具有荧光切换性质的官能团;磷酸分子的数目为4。向反应体系中加入dG4P和dT4P,以及相应的反应缓冲液、酶、金属离子等,发生荧光发生测序反应,产生荧光信号,并被CCD所检测。记录该荧光信号的值。记为第1次反应。将反应残存的dG4P和dT4P洗去。向反应体系中加入dA4P和dC4P,发生和上述相同的测序反应,并记录荧光信号的值。记为第2次反应。该方法也被称为单色2+2测序方法。重复上述过程。凡奇数次加入dG4P和dT4P,凡偶数次加入dA4P和dC4P,得到一组测序信号的值:x=(2,3,3,1,1,3,2,1,2,1)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dC4P和dT4P,凡偶数次加入dA4P和dG4P,得到一组测序信号:y=(1,4,4,2,2,1,1,4,1,1)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dA4P和dT4P,凡偶数次加入dC4P和dG4P,得到一组测序信号:z=(1,1,2,1,4,3,1,3,1,1,2)。将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上三个信号的公共碱基如表3所示:信号xKKMMMKKKMKMMMKKMKKM信号yYRRRRYYYYRRYYRYRRRRYR信号zWSWWSWWWWSSSWSSSWSWW公共碱基TGAA?TTT?G?C?G???GA??表3由于在求三组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第2个值4改为3,同时将信号x的第6个值3改为4,则信号将变为如下表4所示:表4上表中,“信号y的第2个值4改为3”被表示为一个画有删除线的R,“信号x的第6个值3改为4”被表示为增加一个M(以斜体、下划线表示)。经过这两个修改后,三组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。本实施例的短序列,可以有效解释本发明的错误校正的方法。本实施例中,所用的修改方式是修改变动最小的一种,也是使得后面序列匹配的最简单一种方法。实际应用中,可以构建数学模型,简单的实现这种变动。实际可行的算法中,所有可能性的变动都是以概率来统计的,经过概率参数校正以后,上述变动是最可能的正确变动。该计算是基于贝叶斯概型的最大似然法的简单应用。该计算方法为一般的数学方法,属于数学常规知识。所述编码、解码DNA序列的方法,在应用于上述DNA测序信号上时,可以有效提高测序准确率。为了解码,将测序信号表示成一个赋权图(weightedgraph),如图1所示。一个赋权图记为G(V,E,W),其中V为图的节点,E为图的边,W为每条边的权值(实数)。设某个按次计的测序信号为ai。1.对每个信号ai,若第i次呈递的核苷酸为X,则画个ai节点,每个节点代表一个X碱基。2.这ai个节点按顺序顺次、有序相连,即这个节点中的第1个点指向第2个点,第2个点指向第3个点,以此类推。3.这个节点的最后一个节点具有一个指向自己的环。4.表示第i次的所有节点均指向表示第(i+1)次的第一个节点。5.根据大量测序数据的统计结果,给所有的边以权值。如果一条DNA序列分别用M/K、R/Y、W/S三种组合各测了一遍,得到3条测序信号,则将这3条测序信号分别用上述方法表示成图。如图1所示。序列TGAACTTTAGCCACGGAGTA的三组信号分别为:(含错误)M/K:2,3,3,1,1,3,2,1,2,1R/Y:1,4,4,2,2,1,1,4,1,1W/S:1,1,2,1,4,3,1,3,1,1,2定义有向赋权图的路径为:有向赋权图中的一组节点v1v2...vn,这组节点可以全不相同,也可以某些节点相同(例如v1和v2代表相同的节点),且对该组节点中任意相邻的两个节点vi和vi+1该图中均存在一条有向边从vi指向vi+1。定义路径的权值为组成该路径的所有边的权值之和。若将测序信号表示成一个赋权图,则该图中的每一条路径均代表了一种可能的DNA序列。信号解码即找所有图之间的最大公共路径,具体实施的方法有穷举法、贪心法、动态规划法、启发式搜索法等。实施例2根据实施例1上所述的测序方法。对于5000条400bp长的DNA序列进行了解码;每1000条序列为一组,一共分为5组。根据实施例1相同的测序校正方法,将编码的正确率和解码后的正确率统计如下表5所示:编号编码正确率解码后正确率10.97360.991720.98130.995130.98780.997740.99530.999750.99730.9999表5可见本发明所描述的编码-解码方法有效提升了编码过程中的准确率。同时例如当错误率是0.0364的时候,校正后变为0.0083;错误率是0.0047的时候,校正后变为0.0003;相比较,当错误率降低了7.74倍,校正后则降低了27.6倍。总体数据表现出明显的规律:降低测序错误率,会明显导致校正以后数据错误率更加降低。这种规律可以总结为一句话,任何对于测序方法的可以降低错误率的微小改进,都可以更加明显的降低校正以后的编码数据的错误率。分别统计各组的编码准确率和解码后准确率,并用小提琴图和箱型图表示其数值的分布规律(如图2所示)。根据解码中被修改的信号的特征,可以筛选出有较大概率解码正确的序列,进一步提高解码准确性。统计上面数据中每条序列在解码中被修改的信号的数目,其频率分布直方图如图3所示。该频率分布直方图具有如下特征:在图像的左端有一个尖峰,而在该尖峰的右侧呈长尾状分布。如果将下图中处于长尾分布区域的序列丢弃,仅选取处于尖峰区域的序列,则可以进一步将解码后准确率提高2-10倍。图4表示了编码中发生错误的信号的编号和解码中被错误地修改的信号的编号之间的相关关系,横坐标表示编码中发生错误的信号的编号,纵坐标表示解码中被错误地修改的信号的编号之间的相关关系,颜色的灰度表示该点被计数的次数占所有序列中的比例。图3显示在大多数情况下,即使解码中发生了错误,被修改的信号和实际发生错误的信号也相隔非常近。因此,可以利用该特征判断解码的质量。如果某一信号及其邻近的信号均未在解码中被修改,则其所代表的碱基类型具有极高的可信度。实施例3(Illumina)将待测单链DNA分子固定在固相表面上。固定的方法可以是化学交联、分子吸附等,可以把DNA的3’端或者5’端固定。该待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5′-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。向反应体系中加入四种dNTP以及相应的反应缓冲液、酶、金属离子等,每种dNTP的3’端被化学基团封闭,且dGTP和dTTP标记有相同颜色的荧光基团,dATP和dCTP标记有另一种相同颜色的荧光基团。反应中,与待测DNA上待延伸位置上的碱基互补配对的dNTP被DNA聚合酶掺入至DNA新生链上。反应结束后,将残存的dNTP等洗去,用CCD检测并记录荧光信号。重复上述反应,得到一组测序信号的值:x=KKMMMKKKMKMMMKKMKKM。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。重复前述测序过程,但dCTP和dTTP标记相同颜色的荧光基团,dATP和dGTP标记另一种相同颜色的荧光基团。得到另一组测序信号的值:y=YRRRRYYYYRRYYRYRRRRYR。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。重复前述测序过程,但dATP和dTTP标记相同颜色的荧光基团,dCTP和dGTP标记另一种相同颜色的荧光基团。得到另一组测序信号的值:z=WSWWSWWWWSSSWSSSWSWW。将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上三个信号的公共碱基如表6所示:信号xKKMMMKKKMKMMMKKMKKM信号yYRRRRYYYYRRYYRYRRRRYR信号zWSWWSWWWWSSSWSSSWSWW公共碱基TGAA?TTT?G?C?G???GA??表6由于在求三组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第2个值4改为3,同时将信号x的第6个值3改为4,则信号将变为如表7所示:表7上表中,“信号y的第2个值4改为3”被表示为一个画有删除线的R,“信号x的第6个值3改为4”被表示为增加一个M(以斜体、下划线表示)。经过这两个修改后,三组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。实施例4待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5′-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。将待测DNA分子-测序引物复合物分成三份,每份均加入四种dNTP、某几种ddNTP以及DNA合成反应所必需的酶、缓冲液等。所加入的dNTP为天然dNTP,所加入的ddNTP则拥有某种可以被仪器所检测的标记,包括但不限于放射性同位素标记、化学荧光基团标记等。第一份中,ddGTP和ddTTP有相同的标记,ddATP和ddCTP有另一种相同的标记;第二份中,ddCTP和ddTTP有相同的标记,ddATP和ddGTP有另一种相同的标记;第三份中,ddATP和ddTTP有相同的标记,ddCTP和ddGTP有另一种相同的标记。这三份均在适宜条件下反应一段时间,发生DNA合成反应。反应完成后,可以对反应产物进行必要的纯化等处理步骤(也可以不做)。然后对三份反应产物进行DNA电泳实验,根据电泳条带,可以分别获得三条测序信号:x=KKMMMKKKMKMMMKKMKKMy=YRRRRYYYYRRYYRYRRRRYRz=WSWWSWWWWSSSWSSSWSWW将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上三个信号的公共碱基如表8所示:信号xKKMMMKKKMKMMMKKMKKM信号yYRRRRYYYYRRYYRYRRRRYR信号zWSWWSWWWWSSSWSSSWSWW公共碱基TGAA?TTT?G?C?G???GA??表8由于在求三组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第2个值4改为3,同时将信号x的第6个值3改为4,则信号将变为如表9所示:表9上表中,“信号y的第2个值4改为3”被表示为一个画有删除线的R,“信号x的第6个值3改为4”被表示为增加一个M(以斜体、下划线表示)。经过这两个修改后,三组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。实施例5(三轮双色2+2)将待测单链DNA分子固定在固相表面上。固定的方法可以是化学交联、分子吸附等,可以把DNA的3’端或者5’端固定。该待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5’-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。向反应体系中加入dG4P和dT4P(二者标记不同颜色的荧光基团),以及相应的反应缓冲液、酶、金属离子等,发生荧光发生测序反应,产生荧光信号,并被CCD所检测。记录该荧光信号的值。记为第1次反应。将反应残存的dG4P和dT4P洗去。向反应体系中加入dA4P和dC4P(二者标记不同颜色的荧光基团),发生和上述相同的测序反应,并记录荧光信号的值。记为第2次反应。重复上述过程。凡奇数次加入dG4P和dT4P,凡偶数次加入dA4P和dC4P,且每一次加入的两种dN4P均标记不同颜色的荧光基团。得到一组测序信号的值:x=(1G+1T,2A+1C,0G+3T,1A+0C,1G+0T,1A+2C,2G+0T,1A+0C,1G+1T,1A+0C)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dC4P和dT4P,凡偶数次加入dA4P和dG4P,且每一次加入的两种dN4P均标记不同颜色的荧光基团。得到一组测序信号:y=(0C+1T,3A+1G,1C+3T,1A+1G,2C+0T,1A+0G,1C+0T,1A+3G,0C+1T,1A+0G)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dA4P和dT4P,凡偶数次加入dC4P和dG4P,且每一次加入的两种dN4P均标记不同颜色的荧光基团。得到一组测序信号:z=(0A+1T,0C+1G,2A+0T,1C+0G,1A+3T,2C+1G,1A+0T,0C+1G,1A+1T)。该方法可以被称为2+2双色测序,其任意两次的测序数据可以获得基因编码信息;可以认为其是正交的测序结果。将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上三个信号的公共碱基如表10所示:x-AAAAAAAx-CCCCx-GGGGGGx-TTTTTTy-AAAAAAAAy-CCCCCy-GGGGGGy-TTTTTTz-AAAAAAAz-CCCCCz-GGGGGGz-TTTTTT公共碱基TGAA??TT???C?????????表10由于在求三组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第2个值(3A+1G)改为(2A+1G),同时将信号x的第6个值(1A+2C)改为(1A+3C),则信号将变为表11所示:表11上表中,“信号y的第2个值(3A+1G)改为(2A+1G)”被表示为一个画有删除线的A,“信号x的第6个值(1A+2C)改为(1A+3C)”被表示为增加一个C(以斜体、下划线表示)。经过这两个修改后,三组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。实施例6(两轮双色2+2)将待测单链DNA分子固定在固相表面上。固定的方法可以是化学交联、分子吸附等,可以把DNA的3’端或者5’端固定。该待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5’-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。向反应体系中加入dG4P和dT4P(二者标记不同颜色的荧光基团),以及相应的反应缓冲液、酶、金属离子等,发生荧光发生测序反应,产生荧光信号,并被CCD所检测。记录该荧光信号的值。记为第1次反应。将反应残存的dG4P和dT4P洗去。向反应体系中加入dA4P和dC4P(二者标记不同颜色的荧光基团),发生和上述相同的测序反应,并记录荧光信号的值。记为第2次反应。重复上述过程。凡奇数次加入dG4P和dT4P,凡偶数次加入dA4P和dC4P,且每一次加入的两种dN4P均标记不同颜色的荧光基团。得到一组测序信号的值:x=(1G+1T,2A+1C,0G+3T,1A+0C,1G+0T,1A+2C,2G+0T,1A+0C,1G+1T,1A+0C)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dC4P和dT4P,凡偶数次加入dA4P和dG4P,且每一次加入的两种dN4P均标记不同颜色的荧光基团。得到一组测序信号:y=(0C+1T,3A+1G,1C+3T,1A+1G,2C+0T,1A+0G,1C+0T,1A+3G,0C+1T,1A+0G)。将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上两个信号的公共碱基是表12所示:表12由于在求两组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第2个值(3A+1G)改为(2A+1G),同时将信号x的第6个值(1A+2C)改为(1A+3C),则信号将变为表13所示:表13上表中,“信号y的第2个值(3A+1G)改为(2A+1G)”被表示为一个画有删除线的A,“信号x的第6个值(1A+2C)改为(1A+3C)”被表示为增加一个C(以斜体、下划线表示)。经过这两个修改后,两组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。实施例7(单色1+3)将待测单链DNA分子固定在固相表面上。固定的方法可以是化学交联、分子吸附等,可以把DNA的3’端或者5’端固定。该待测DNA包含一段序列已知的固定片段,可以和测序引物互补杂交。从该固定片段的3’端开始至该待测DNA分子的3’端的区域为待测区域。在该实例中,待测区域的序列为5’-TGAACTTTAGCCACGGAGTA-3’。将测序引物杂交到待测DNA分子的固定片段区域。向反应体系中加入dC4P、dG4P和dT4P,以及相应的反应缓冲液、酶、金属离子等,发生荧光发生测序反应,产生荧光信号,并被CCD所检测。记录该荧光信号的值。记为第1次反应。将反应残存的dC4P、dG4P和dT4P洗去。向反应体系中加入dA4P,发生和上述相同的测序反应,并记录荧光信号的值。记为第2次反应。重复上述过程。凡奇数次加入dC4P、dG4P和dT4P,凡偶数次加入dA4P。得到一组测序信号的值:x=(2,2,4,1,3,1,3,1,2,1)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dA4P、dG4P和dT4P,凡偶数次加入dC4P。得到一组测序信号:y=(4,1,6,2,1,1,6)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dA4P、dC4P和dT4P,凡偶数次加入dG4P。得到一组测序信号:z=(1,1,7,1,4,2,1,1,2)。利用高温或强亲水物质(如尿素、甲酰胺等)将上述测序反应中合成的DNA新生链解链并洗去。重新杂交测序引物。凡奇数次加入dT4P,凡偶数次加入dA4P、dC4P和dG4P。得到一组测序信号:w=(1,4,3,9,1,1)。将上述测序信号的值和其所代表的碱基类型结合起来,得到的按位表示的测序信号及每个位置上四个信号的公共碱基如表14所示:信号xBBAABBBBABBBABBBABBA信号yDDDDCDDDDDDCCDCDDDDDD信号zHGHHHHHHHGHHHHGGHGHH信号wTVVVVTTTVVVVVVVVVTV公共碱基TGAACTTTAG?C???GA????表14由于在求四组信号在每个位置上的公共碱基时发现,若干位置上均无公共碱基,因此断定出现测序错误。将信号y的第3个值6改为5,同时将信号w的第4个值9改为10,则信号将变为如表15所示:表15上表中,“信号y的第3个值6改为5”被表示为一个画有删除线的D,“信号w的第4个值9改为10”被表示为增加一个V(以斜体、下划线表示)。经过这两个修改后,四组信号的所有位置上均有公共碱基,且这些公共碱基组成的序列恰好为待测DNA的序列,表明这种编码DNA序列的方法有效检测出测序过程中发生的错误,而解码方法有效地将这些错误校正。本发明中所用到的具体的测序方法均为本领域的常规技术。实施例1所用到的方法是常规的举例,并没有针对序列的强制限定或者要求。实施例1是为了表达本发明的发明点的一个常规举例。本发明中实施例并未列出所有的适用测序方法和范围;本领域技术人员可以根据本发明的指引,对测序方法进行组合。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1