一种高度相似微生物的鉴定和分类方法与流程
本发明属于微生物鉴定与分类技术领域,更具体地说,涉及通过计算不同微生物菌株的基因组所包含基因内容之间的非相似性,将高度近似的微生物进行鉴定与分类的方法。
背景技术:
微生物的鉴定与分类是微生物学研究中的基本问题。传统方法主要通过显微镜观察微生物的形态学特征,并设计相关实验测量微生物的生理生化参数,以此判断微生物所属种类。然而该类方法实验复杂,缺少定量标准,且不同实验人员及不同实验室所测得参数不尽相同。所以,微生物学科研人员致力于寻找实验步骤简单且具有量化标准的方法对微生物菌株进行鉴定与分类。
在过去几十年,多种方法被开发用于微生物的快速鉴定与分类。其中,dna-dna杂交(ddh)和16srrna基因相似性是应用最广泛的两种方法。其中,70%的ddh杂交率至今为止还是微生物物种鉴定的黄金标准。然而,ddh实验步骤相对复杂,在不同实验室之间难以标准化,且ddh的值随模板dna与探针dna交换而发生改变。因此,目前16srrna基因相似性已逐步取代ddh作为物种鉴定的新标准。其中,微生物学界广泛接受97%的16srrna基因相似性作为两个菌株属于同一个物种的标准参数,且ddh仅需要在16srrna基因相似性达到97%的时候进行。
在后基因组时代,随着微生物基因组不断地被测序,多种基于基因组手段的微生物鉴定与分类方法被开发,包括计算机模拟ddh,平均氨基酸相似性,平均核酸相似性,以及多位点序列分析。由于整合了基因组信息,这些方法在准确度和可靠度上基本超过了16srrna基因相似性。其中,94%~96%的平均核酸相似性和平均氨基酸相似性在微生物物种鉴定上与70%的ddh和97%的16srrna基因相似性高度对于,并已逐渐成为后基因组时代微生物物种鉴定的黄金标准。
然而,针对高度近似微生物的鉴定与分类问题,现有的方法并不能有效解决问题,因为不管16srrna基因还是平均核酸/氨基酸相似性对于高度近似菌都极其一致,很难区分。
技术实现要素:
本发明的目的在于:克服现有方法无法准确鉴定和分类高度相似微生物等问题,本发明的发明人发现对于高度近似菌,微生物菌株分化的主要驱动因子在于新型功能基因的获得与丢失,而非单核苷酸位点突变,提供一种准确的高度相似微生物的鉴定和分类方法。
为了实现上述发明目的,本发明提供一种高度相似微生物的鉴定和分类方法,其包括如下步骤:
(1)使用eggnog数据库对微生物基因组编码的蛋白序列进行比对,赋予每个微生物基因组一个eggnog基因id,生成一个基于eggnog数据库的微生物基因谱;
(2)计算基因谱中不同待测微生物之间的基因内容非相似性,得到一个非相似性矩阵;
(3)对矩阵进行聚类分析,将基因内容类似的微生物聚为一簇,判定为同一类微生物,完成微生物的鉴定和分类。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,所述过滤除菌是使用0.22μm滤膜过滤。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,步骤(1)中,所述肠道内容物样品为0.3~0.5g。可见,本发明前处理方法仅需少量样品即可完成。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,步骤(1)中,所述基因谱的横向为eggnog基因id,纵向为微生物。
更进一步地,所述基因谱的构建步骤如下:
(a)提取微生物基因组中所有基因编码的蛋白序列;
(b)将蛋白序列与eggnog数据库进行类blast比对,使用参数为e值1e-5,全局序列相似性30%;
(c)将比对结果中最好的eggnog基因id赋予目标基因,最后生成一张每行代表不同eggnog基因,每纵列代表不同微生物的表。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,步骤(2)中,所述矩阵的纵向和横向均为微生物,矩阵中的值代表非相似性。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,步骤(2)中,采用bray-curtis非相似性算法计算。
作为本发明高度相似微生物的鉴定和分类方法的一种优选技术方案,可采用pca聚类,将多维矩阵降维至二维进行分析,功能类似的微生物将聚在一起。
与现有技术相比,本发明具有如下有益效果:
本发明方法针对高度相似微生物,创造性地提出微生物水平差异主要驱动因子为基因的获得与丢失,而非传统手段常考虑的核酸位点突变。本发明对高度相似微生物的鉴定和分类有极高的准确性,具有良好的应用前景。
附图说明
下面结合附图和具体实施方式,对本发明及有益效果进行详细说明。
图1是本发明鉴定和分类方法示意图。
图2是本发明实施例1鉴定分类结果示意图。
图3是本发明实施例2鉴定分类结果示意图。
具体实施方式
为了使本发明的发明目的、技术方案和有益技术效果更加清晰,以下结合实施例,对本发明进行进一步详细说明。应当理解的是,本说明书中描述的实施例仅仅是为了解释本发明,并非为了限定本发明,实施例的配方、比例等可因地制宜做出选择而对结果并无实质性影响。
实施例1肠杆菌的鉴定与分类
1)菌株收集
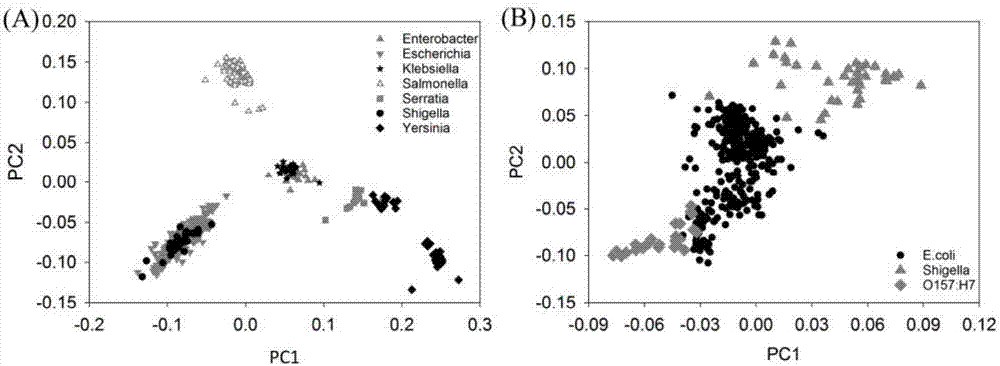
本例一共收集已测序的916株肠杆菌基因组,其中肠杆菌属14株,埃希氏菌属384株,克雷白氏杆菌属45株,沙门氏菌属314株,沙雷氏菌属14株,志贺氏菌属42株,耶尔森氏鼠疫杆菌103株。
2)菌株鉴定与分类
当把所有菌属混在一起进行鉴定分类工作时,大多数菌属可以较好的分类。但是其中志贺氏菌属和埃希氏菌属很难区分,肠杆菌属与克雷白氏杆菌属也较难区分但是趋势明显。然而,耶尔森氏鼠疫杆菌中y.pestis和y.pseudotuberculosis与其它耶尔森氏鼠疫杆菌区分度明显。这些现象与目前临床所观察到的结论基本一致。进一步对埃希氏菌属和志贺氏菌属单独聚类分析发现,该两类菌属在高分辨率下区分度明显。同时,埃希氏菌属o157系列与其它埃希氏菌属区分度亦非常明显,说明志贺氏菌属和o157系列埃希氏菌可作为埃希氏菌属下不同的物种对待。该例子解决了当前肠杆菌鉴定分类中所面临的不少难题。
实施例2链球菌的鉴定与分类
1)菌株收集
本例共收集283株链球菌菌株,包括83株s.agalactiae,7株s.mitis,8株s.oralis,38株s.mutans,119株s.pneumoniae,12株s.pyogenes,10株s.suis,以及6株s.thermophilus。
2)菌株鉴定与分类
对该283株链球菌菌株聚类分析,所有物种都能很好的进行区分,包括s.oralis和s.mitis。s.oralis和s.mitis在微生物分类学上高度相似,一度认为是同一个物种单元。而基于基因内容非相似性聚类,该2类链球菌存在很明显的区分度,可认为属于不同的微生物物种。
- 还没有人留言评论。精彩留言会获得点赞!