一种引入序列信息的残基相互作用网络比对算法SI‑MAGNA的制作方法
本发明是一种引入序列信息的残基相互作用网络比对算法si-magna,属于计算机应用技术领域。具体的说就是基于magna网络比对方法,提出引入蛋白质序列信息的残基相互作用网络比对算法si-magna,发现两个蛋白质结构上的保守区域,从而找到实现相似功能的相似结构和产生差异的独有结构,该方法可应用于分子设计、分子筛选、药物设计等诸多领域。
背景技术:
随着实验测定技术的发展,产生了大量的分子相互作用数据,也称为生物网络数据,例如:蛋白质-蛋白质相互作用网络、代谢网络、残基相互作用网络、基因表达网络等。这使得生物网络比对在近年来成为研究代谢、结构、功能和进化的一类重要的方法。残基相互作用网络对于从系统角度研究蛋白质空间结构和蛋白质性质、功能的关系有着至关重要的作用。而残基相互作用网络比对对于研究蛋白质的分子基础和空间结构非常重要,它能够推动蛋白质结构、性质和功能相关研究的发展。
目前,绝大多数的网络比对方法是针对蛋白质-蛋白质相互作用网络提出的。kuchaiev,o(《integrativenetworkalignmentrevealslargeregionsofglobalnetworksimilarityinyeastandhuman》,bioinformatics,2011,27(10):1390–1396)等人整合了网络节点之间的多种类型的相似性标准,并决定了相似性标准间的权重,利用最大权重双边图找出最优比对。然而该方法整合了基于蛋白质生物信息的相似性标准,对于残基相互作用网络比对,需要在比对时排除这一因素,比对过程比较复杂。maoguogong(《globalbiologicalnetworkalignmentbyusingefficientmemeticalgorithm》,ieee/acmtransactionsoncomputationalbiologyandbioinformatics,2016,13(6):1117-1129)等人将遗传算法与局部搜索相结合,首先使用一个粗略的相似度得分矩阵进行初始化,然后使用特定的邻域启发式局部搜索策略来找到最优比对。然而该方法针对蛋白质-蛋白质相互作用网络比对,它需要蛋白质的节点信息作为比对的优化条件,对于残基相互作用网络比对,无法产生在网络拓扑和一级结构方面更准确的比对结果。somayehashemifar(《modulealign:module-basedglobalalignmentofprotein–proteininteractionnetworks》,bioinformatics,2016,32(17):i658–i664)等人利用局部信息来定义模块的同源性分数,基于参与相同模块的功能相关蛋白质的分层聚类,并采用迭代方案找到两个网络之间的比对。然而该方法针对蛋白质-蛋白质相互作用网络,需要蛋白质同源性方面的信息作为比对的重要依据,对于残基相互作用网络比对,无法产生在生物方面合理的比对结果。
生物网络比对是一种通过比较两个或多个相互作用网络,发现不同网络在拓扑和功能上的相似区域的方法,已经在研究生物分子的结构和功能,分析生物的进化和演变等领域有了重要应用。然而文献及专利中未见有针对于残基相互作用网络比对的算法,更未见将蛋白质的序列信息引入到残基相互作用网络比对中的算法。
技术实现要素:
有鉴于此,本发明的目的是将蛋白质的序列信息引入到网络比对算法的优化函数中,以实现在残基相互作用网络比对中,产生在网络拓扑和一级结构方面更准确更合理的比对结果。
本发明的技术方案:一种引入序列信息的残基相互作用网络比对算si-magna,采用下列步骤:
(1)引入蛋白质的序列信息相似性得分
blosum矩阵是一种通过统计相似蛋白质序列的替换率而得到的氨基酸替换矩阵。blosum矩阵基于蛋白质进化的星状模型(即忽略物种近端和远端的关系)和区块中的保守位置与置换关系进行计分,这对于发现同源蛋白质中的保守区域有非常重要的作用。同时,这也与残基相互作用网络比对发现和探索不同网络间保守区域和差异的目标一致。因此,以blosum矩阵作为蛋白质的序列比对的打分矩阵,将比对后残基的序列相似性得分信息加入到残基相互作用网络比对方法中。
(2)构建适应度函数f
适应度函数f作为网络比对的优化条件在算法中起到至关重要的作用,将适应度函数f定义为:
f=α×toposcore(f)+(1-α)×seqscore(f),α∈[0,1]
其中toposcore(f)表示拓扑信息的相似性,seqscore(f)表示序列信息的相似性,权重α用来调整拓扑信息相似性和序列信息相似性在适应度函数中所占的比例,α∈[0,1]。
拓扑信息的相似性toposcore可以从现有的3种拓扑比对质量评价标准中选择,包括:边正确性得分(ec),诱导保守结构得分(ics)和对称子结构得分(s3)。
由网络g1(v1,e1)和g2(v2,e2)的比对f:v1→v2,设g2[y]为点集为y的g2的子网,f(v1)={f(v)∈v2:v∈v1},f(e1)={(f(u),f(v))∈e2:(u,v)∈e1},并将保守边定义为由通过f比对的两个网络的两条边组成。
边正确性得分(ec)表示比对中保守边的数量占网络g1边的数量的比例。当一个输入网络g1与另一网络g2是同构的时,它取得最高值100%。ec可表示为:
诱导保守结构得分(ics)表示保守边的数量占通过比对f形成的网络g2子网的边的数量的比例,表示为:
对称子结构得分(s3)表示保守边的数量占网络g1和g2[f(v1)]叠加的复合图边的数量的比例,表示为:
序列信息的相似性seqscore使用基于blosum打分矩阵进行序列比对后残基的序列相似性得分信息。
通过调节拓扑信息-序列信息权重α的大小,考察拓扑信息相似性和序列信息相似性所占的比例对比对结果的影响,以获得更加优秀更加合理的比对结果。拓扑信息-序列信息权重α值的取值范围是[0,1],当α值等于1时,表示只引入拓扑信息相似性,而不考虑序列信息相似性;当α等于0时,表示只引入序列信息相似性,而不考虑拓扑信息相似性。
(3)基于遗传算法的框架进行网络比对
本方法基于遗传算法框架,迭代搜索以获得比对结果。通过随机比对以获得给定种群规模p的初始种群p0,种群中的成员即比对。对于每一代种群p,通过适应度函数f将种群中的成员以其比对质量从高到低排序,设定精英率e,将种群中占比为e的高适应度成员保留作为子代成员直接加入下一代种群中。使用roulette选择算法选择种群p中的成员进行“交叉”产生新的子代以补足下一代中剩余的部分,所选择成员的概率与成员的适应度成正比。种群中成员被选中的概率可表示为:
算法si-magna具体步骤如下:
步骤1:输入网络g1、g2及相关参数:代数n、精英率e、种群规模p;
步骤2:随机产生种群规模p的比对初始种群p0;
步骤3:以初始种群p0作为父代种群;
步骤4:设置代数计数器n=1;
步骤5:计算父代种群p中成员的适应度值,并进行排序。
步骤6:判断并保留适应度值高的父代种群成员,保留比例为精英率e,即p′1=p·e。
步骤7:通过roulette选择算法和交叉函数产生适应度值较高的比对成员,补足剩余的部分,即p′2=p·(1-e)。
步骤8:将(5)(6)步骤产生的比对成员组成子代成员p’;
步骤9:当n达到代数n时,终止循环;
步骤10:输出网络比对结果。
本发明的有益效果:本发明公开了一种引入序列信息的残基相互作用网络比对算法si-magna。本发明方法是针对magna算法在残基相互作用网络比对方面精确度不高,比对结果不理想的缺陷做出改进,使改进后的si-magna算法在残基相互作用网络比对方面的比对结果更加精确,具有更高的边正确性ec。si-magna算法以blosum矩阵作为蛋白质的序列比对的打分矩阵,将比对后残基的序列相似性得分信息加入到网络比对的优化函数中,使比对过程受到网络拓扑和序列信息两方面因素的影响,提高了比对结果在网络拓扑和一级结构上的准确性和合理性。本发明方法在magna算法的基础上,针对该算法的一些缺陷做出改进,为从系统角度探索蛋白质结构对蛋白质性质、功能的影响提供了一个新的途径。
具体实施方式
(1)本文选择3组残基相互作用网络对作为算法的输入,分别是:(a)来自嗜热栖热菌的adp核糖焦磷酸酶(pdb号:1v8i)和来自结核分枝杆菌的结构水解酶(pdb号:1mp2);(b)来自高纬度温带海域鱼类的ⅲ型抗冻蛋白质异构体(pdb号:9ame)和来自人类的唾液酸合成酶(pdb号:1wvo);(c)来自枯草芽孢杆菌的野生嗜温型脂肪酶(pdb号:1i6w)和其两个嗜热型突变体(pdb号:3d2b和3qmm)。不同蛋白质对的三维结构信息和序列信息从rcsbpdb数据库(http://www.rcsb.org/pdb/home/home.do)中获得,采用blast算法进行序列比对;并计算残基-残基的相互作用,以此来构建残基相互作用网络,然后进行网络比对。
(2)使用si-magna算法对3组残基相互作用网络对进行网络比对。拓扑信息-序列信息权重α值是影响比对结果的重要因子,它的取值范围是[0,1]。在实验中,设定代数n为2000,α取值步长为0.1。实验结果如附图说明中图1所示。
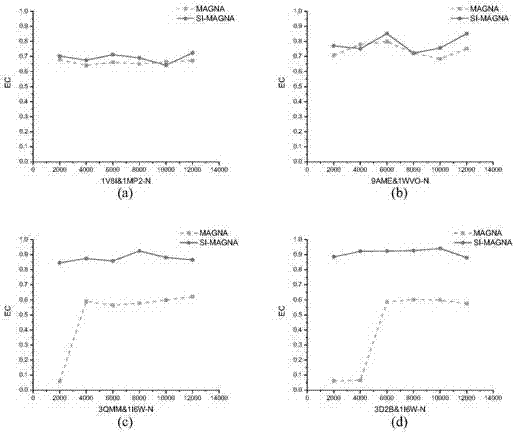
(3)si-magna算法与magna算法的网络比对结果比较。拓扑信息-序列信息权重α取各组在n=2000时产生最优比对时的值,代数n取值为2000、4000、6000、8000、10000、120000。实验结果如附图说明中图2所示。
(4)si-magna算法与适用于残基相互作用网络比对的其他方法(graal、mi-graal和cytogedevo算法)相比较,实验结果如附图说明中图3所示。
附图说明:
图1:si-magna算法中拓扑信息-序列信息权重α对比对结果的影响。
图2:si-magna算法与magna算法的网络比对结果比较。
图3:si-magna算法与graal、mi-graal和cytogedevo算法的网络比对结果比较。
本方法基于magna网络比对方法,将蛋白质的序列信息作为序列信息相似性引入到优化函数中,提出更加适用于残基相互作用网络比对的si-magna算法。由图1可见,在残基相互作用网络的比对中,仅依靠网络的拓扑信息或序列信息均不能获得很好的比对结果,引入序列信息相似性能够明显提升比对质量,获得更优的比对结果。由图2可见,相对于magna算法,si-magna算法不但取得了更优的比对结果,同时因为更少的代数提高了比对的效率。由图3可见,由于考虑了蛋白质序列信息的影响,加入了序列信息相似性特征,使si-magna算法比graal、mi-graal和cytogedevo算法的比对结果具有更高的边正确性ec,比对结果在网络拓扑和一级结构上更加准确合理。
- 还没有人留言评论。精彩留言会获得点赞!