包含RAF抑制剂和ERK抑制剂的治疗组合的制作方法
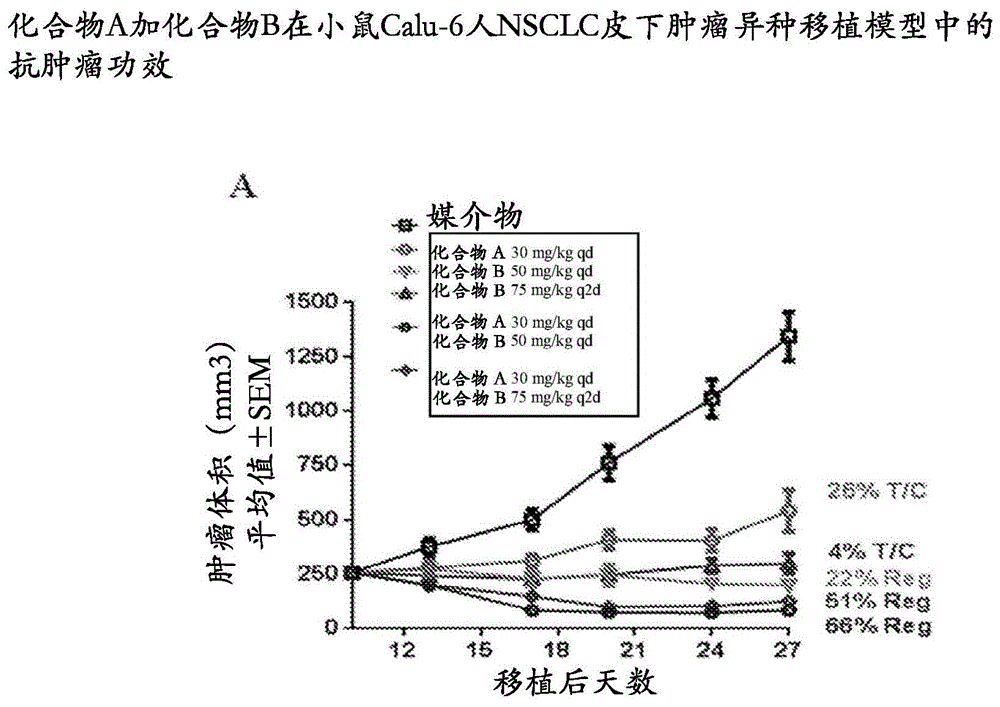
本发明涉及raf抑制剂、特别地c-raf(c-raf或craf)的抑制剂用于治疗癌症的用途,该癌症是具有丝裂原活化蛋白激酶(mapk)改变的晚期实体瘤,如kras-突变型肿瘤,并且特别地是kras-突变型nsclc(非小细胞肺癌)、黑素瘤、胰腺癌、结肠直肠癌和卵巢癌。本发明特别涉及使用至少一种raf抑制剂用于治疗癌症的治疗组合。本发明还涉及erk抑制剂(erki)用于治疗癌症(具体地具有kras-突变、以及特别地kras的功能获得性突变的癌症)的用途,这些癌症包括肺癌(具体地nsclc)、黑素瘤、胰腺癌、和卵巢癌。本发明进一步涉及药物组合,该药物组合包含(a)至少一种erk抑制剂(erki)、和(b)raf抑制剂(其优选为也可抑制b-raf的c-raf(craf)抑制剂),其中制备和/或使用这两种化合物用于同时、分开或顺序给药用于治疗增殖性疾病,并且本发明涉及包含此类组合的药物组合物;治疗患有增殖性疾病的受试者的方法,该方法包括将所述组合给予对其有需要的受试者;这种组合用于治疗增殖性疾病的用途;以及包含这种组合的商业包装。在本发明中,所述增殖性疾病通常是具有丝裂原活化蛋白激酶(mapk)改变的实体瘤,如kras-突变型肿瘤,并特别地是kras-突变型nsclc(非小细胞肺癌)、黑素瘤、胰腺癌、结肠直肠癌和卵巢癌。通常,craf抑制剂和erk抑制剂都是低分子量化合物,并特别地,本发明涉及用于本文所述的用途的化合物a和化合物b的组合。特别地,提供了包含化合物a、或其药学上可接受的盐,以及化合物b、或其药学上可接受的盐的药物组合。此药物组合可以特别地有用于治疗kras或braf突变型nsclc(包括晚期或转移性kras或braf突变型nsclc)。
背景技术:
:ras/raf/mek/erk或mapk途径是驱动细胞增殖、分化和存活的关键信号传导级联。该途径的失调是许多肿瘤发生实例的基础。mapk途径的异常信号传导或不当活化已显示于多种肿瘤类型(包括黑素瘤、肺癌和胰腺癌)中,并且可以通过几种不同的机制发生,包括活化ras和braf中的突变。ras是gtp酶的超家族,并且包括kras(v-ki-ras2kirsten大鼠肉瘤病毒致癌基因同源物),它是受调节的信号传导蛋白,可以由各种单点突变发动(活化),这被称为功能获得性突变。mapk途径频繁在人癌症中发生突变,其中kras和braf突变是最常见的(近似30%)。ras突变,特别是功能获得性突变,已在9%-30%的所有癌症中检测到,其中kras突变的患病率最高(86%),其次是nras(11%),而且很少发生的是hras(3%)(coxad,fesiksw,kimmelmanac等人(2014),natrevdrugdiscov.[自然评论药物发现]11月;13(11):828-51)。虽然选择性braf抑制剂(brafi),以及在较小程度上,mek抑制剂(meki)已证明在braf突变型肿瘤中具有良好活性,但目前尚无针对kras突变型肿瘤的有效疗法(cantwell-dorriser,o'learyjj,sheilsom(2011)molcancerther.[分子癌症治疗学]3月;10(3):385-94.)。关于craf在介导kras信号传导和kras突变型非小细胞肺癌(nsclc)发展中发挥作用的新证据使其成为治疗干预的合适靶标(blascorb,francozs,santamaríad等人(2011)c-raf,butnotb-raf,isessentialfordevelopmentofk-rasoncogene-drivennon-smallcelllungcarcinoma[c-raf而不是b-raf对于k-ras致癌基因驱动的非小细胞肺癌的发展至关重要].cancercell.[癌细胞]2011年5月17日;19(5):652-63.)。据显示,在kras突变型癌症中,在用meki治疗后,craf促进反馈介导的途径再活化(litop,saborowskia,yuej等人(2014)disruptionofc-raf-mediatedmekactivationisrequiredforeffectivemekinhibitioninkrasmutanttumors.[破坏craf介导的mek活化对kras突变型肿瘤中有效的mek抑制是所需的]cancercell[癌细胞]25,697-710.,lamba等人2014)。此外,craf在介导brafi治疗后的反常活化中发挥重要作用(poulikakospi,zhangc,bollagg等人(2010),nature[自然].mar18;464(7287):427-30.,hatzivassiliou等人2010,heidorn等人2010)。因此,有效抑制craf和braf活性的选择性泛raf抑制剂可有效阻断braf突变型肿瘤和ras突变体驱动的肿瘤发生,并且还可减少反馈活化。本文所述的化合物a是craf和braf的有效抑制剂。肺癌是影响全球男性和女性的常见癌症。nsclc是最常见的肺癌类型(大致85%),其中近似70%的患者在诊断时呈现出疾病晚期(iiib期或iv期)。约30%的nsclc肿瘤含有活化型kras突变,并且这些突变与对egfr酪氨酸激酶抑制剂(tki)的抗性相关(paow,wangty,rielygj等人(2005)plosmed[公共科学图书馆医学];2(1):e17)。还在黑素瘤(britishj.cancer[英国癌症杂志]112,217-26(2015))、胰腺癌(gastroenterologyvol.[胃肠病学卷]144(6),1220-29(2013))和卵巢癌(britishj.cancer[英国癌症杂志]99(12),2020-28(2008))中发现了活化型kras突变。在高达3%的nsclc中观察到braf突变,并且还被描述为egfr突变阳性nsclc中的抗性机制。已证明直接抑制kras是具有挑战性的。迄今为止,没有经过批准的靶向治疗可用于患有kras突变型癌症(如nsclc)的患者。因此,患有kras突变型nsclc的患者和患有braf突变型nsclc的患者存在高度未满足的医疗需求。因此需要安全和/或良好耐受的靶向疗法。在临床环境中将产生持久和持续应答的疗法是有益的。技术实现要素:本发明还提供了药物组合,该药物组合包含(a)作为化合物a的craf抑制剂、或其药学上可接受的盐,以及(b)作为化合物b的erk抑制剂、或其药学上可接受的盐。这种组合在本文中称为“本发明的组合”。本发明进一步提供了用于同时、分开或顺序(以任何顺序)给予的药物组合,用于在增殖性疾病中使用,该药物组合包含如本文所述作为化合物a的c-raf激酶抑制剂或其药学上可接受的盐和作为化合物b的erk抑制剂或其药学上可接受的盐。本发明特别涉及用于在增殖性疾病的治疗中使用的本发明组合,其特征在于活化mapk途径中的突变,并且特别是kras或braf中的一个或多个突变。本发明特别涉及kras-突变型nsclc(非小细胞肺癌)、braf-突变型nsclc、kras-突变型胰腺癌、kras-突变型结肠直肠癌(crc)、和kras-突变型卵巢癌的治疗。化合物a是braf(本文中也称为b-raf或b-raf)和craf(本文中也称为c-raf或c-raf)蛋白激酶的三磷酸腺苷(atp)竞争性抑制剂。贯穿本披露,化合物a也称为c-raf(或craf)抑制剂或c-raf/c-raf激酶抑制剂。化合物a是n-(3-(2-(2-羟基乙氧基)-6-吗啉代吡啶-4-基)-4-甲基苯基)-2-(三氟甲基)异烟酰胺,并且是具有以下结构的化合物:在基于细胞的测定中,化合物a在细胞系中证明了抗增殖活性,所述细胞系含有活化mapk信号传导的多种突变。在体内,用化合物a治疗在若干种kras突变型模型中产生肿瘤消退,这些kras突变型模型包括nsclc衍生的calu-6(krasq61k)和nci-h358(krasg12c)。总体而言,以良好耐受的剂量观察到的化合物a的体外和体内mapk-途径抑制和抗增殖活性表明,化合物a在肿瘤(该肿瘤在mapk途径中具有活化病变)患者中可以具有抗肿瘤活性。此外,化合物a是b-raf和c-raf两者的2型atp竞争性抑制剂,其使激酶袋保持无活性构象,从而减少许多b-raf抑制剂所见的反常活化,并阻断突变体ras驱动信号传导和细胞增殖。化合物a在许多mapk驱动的人癌细胞系和异种移植肿瘤中表现出功效,该异种移植肿瘤代表在kras、nras和braf癌基因方面具有人病变的模型肿瘤。化合物b是细胞外信号调节激酶1和2(erk1/2)的抑制剂。化合物b通过4-(3-氨基-6-((1s,3s,4s)-3-氟-4-羟基环己基)吡嗪-2-基)-n-((s)-1-(3-溴-5-氟苯基)-2-(甲基氨基)乙基)-2-氟苯甲酰胺的名称已知,并且是以下结构的化合物。已经显示化合物b在各种实体瘤的模型中作为单一药剂治疗具有活性,并且当与第二抗癌治疗剂组合使用时特别有效。例如,在胰管腺癌(pdac,特别难以治疗的癌症(根据cancers[癌症],(basel),第8(4)卷,45(2016年4月)),pdac的5年生存率为7%))模型中,化合物b与若干种不同的抗癌剂组合显示出显著的肿瘤收缩,并且比基于某些组合的组分的单一药剂活性的预期更有效。特别地,与人nsclc异种移植模型calu-6中的单一药剂疗法相比,化合物b与化合物a的组合证明了肿瘤应答的深度和持久性增加。因此,结合泛raf抑制剂(如化合物a)与erk1/2激酶抑制剂(如化合物b)的垂直mapk(丝裂原活化蛋白激酶)抑制预期将优化kras和braf突变型nsclc中mapk信号传导的抑制。此组合还可以有助于防止brafv600e-突变型nsclc中braf和mek(丝裂原活化蛋白激酶)抑制剂组合的出现。因此,本发明提供了使用与raf抑制剂(并特别地与化合物a)组合的化合物b的组合物和方法,用于治疗实体瘤,特别地表达kras突变(包括nsclc,并且具体地kras-突变型nsclc,以及braf突变体nscl(包括brafv600e-突变型nsclc))的肿瘤。本文披露的是药物组合,其包含(a)c-raf抑制剂(如化合物a、或其药学上可接受的盐)、和(b)erk抑制剂(如化合物b、或其药学上可接受的盐),用于同时、分开或顺序给予,用于治疗增殖性疾病,特别地具有丝裂原活化蛋白激酶(mapk)改变的实体瘤,如kras-突变型肿瘤、和某些braf-突变型肿瘤。这些包括kras-突变型nsclc(非小细胞肺癌)、braf-突变型nsclc(非小细胞肺癌)、kras-突变型和braf-突变型nsclc(非小细胞肺癌)、kras-突变型胰腺癌、kras-突变型结肠直肠癌(crc)和kras-突变型卵巢癌。本发明还提供了本发明的药物组合,用于治疗brafv600-突变型黑素瘤,包括复发或难治的brafv600-突变型黑素瘤。还披露了包含此类组合的药物组合物;治疗患有增殖性疾病的受试者的方法,该方法包括将此类组合向对其有需要的受试者给予;这种组合用于治疗增殖性疾病的用途;以及包含这种组合的商业包装。本发明的其他特征、目的和优点会从说明书和附图以及权利要求书中显而易见。附图说明图1a描述了化合物a和化合物b在小鼠中的calu-6nsclc异种移植肿瘤模型中分开或一起使用的功效。如所示,每日(qd)或每隔一日(q2d)口服给予化合物。图1b描绘了图1a中所示治疗的响应持久性,表明组合治疗优于单一试剂治疗。图1c描绘了图1a中的治疗的持久性,如体重随时间的变化所示。具体实施方式本发明的药物组合是化合物a或其药学上可接受的盐、以及化合物b或其药学上可接受的盐的药物组合。在优选的实施例中,提供了本发明的药物组合,用于治疗选自以下的增殖性疾病:kras-突变型nsclc(非小细胞肺癌)、braf-突变型nsclc(包括brafbrafv600e-突变型nsclc)、kras-突变型胰腺癌、kras-突变型结肠直肠癌(crc)和kras-突变型卵巢癌。可能从药物组合中受益的患者包括患有晚期或转移性疾病的患者,例如nsclc患者,诊断为晚期或转移性kras或braf突变型nsclc,在接受标准治疗后可能已经有进展。craf激酶抑制剂本发明的craf激酶抑制剂包括化合物a。化合物a在wo2014/151616中作为实例1156披露。wo2014/151616还描述了包含化合物a的其制备和药物组合物。在本文所述方法、治疗、组合和组合物的优选实施例中,craf抑制剂是化合物a或其药学上可接受的盐。化合物a具有以下结构:化合物a(化合物a)也称为n-(3-(2-(2-羟基乙氧基)-6-吗啉代吡啶-4-基)-4-甲基苯基)-2-(三氟甲基)异烟酰胺。在基于细胞的测定中,化合物a在细胞系中证明了抗增殖活性,所述细胞系含有活化mapk信号传导的多种突变。在体内,用化合物a治疗在若干种kras突变型模型中产生肿瘤消退,这些kras突变型模型包括nsclc衍生的calu-6(krasq61k)和nci-h358(krasg12c)。总体而言,以良好耐受的剂量观察到的化合物a的体外和体内mapk-途径抑制和抗增殖活性表明,化合物a在肿瘤(该肿瘤在mapk途径中具有活化病变)患者中可以具有抗肿瘤活性。此外,化合物a是b-raf和c-raf两者的2型atp竞争性抑制剂,其使激酶袋保持无活性构象,从而减少许多b-raf抑制剂所见的反常活化,并阻断突变体ras驱动信号传导和细胞增殖。化合物a在许多mapk驱动的人癌细胞系和异种移植肿瘤中表现出功效,该异种移植肿瘤代表在kras、nras和braf癌基因方面具有人病变的模型肿瘤。基于化合物a的作用机制,关于craf在mapk途径调节中的重要性的临床前数据和公开文献,化合物a与mapk途径的至少一种其他抑制剂如化合物b组合有途径用于治疗患有mapk改变的晚期实体瘤患者。化合物a可用于在需要此类治疗受试者中治疗(例如,减少、抑制或延迟进展中的一种或多种)增殖性疾病,其是具有丝裂原活化蛋白激酶(mapk)改变的晚期实体瘤,如kras-突变型肿瘤,并且特别地,表达至少一种ras或raf的功能获得性突变的肿瘤,包括肺癌、nsclc(非小细胞肺癌)、卵巢癌、胰腺癌、结肠直肠癌或黑素瘤。erk抑制剂用于本发明的erki化合物通常是化合物b,处于游离形式或药学上可接受的盐。化合物b是细胞外信号调节激酶1和2(erk1/2)的抑制剂。披露了此化合物及其制备,并且包含此化合物的药物组合物描述在公开的pct专利申请wo2015/066188中。化合物b具有以下结构:在一些实施例中,使用化合物b的盐酸盐。治疗用途在一个实施例中,本发明特征在于治疗(例如,抑制、减轻、改善或预防)受试者中的障碍,例如过度增殖性病症或障碍(例如,癌症)的方法。该方法包括向受试者给予与c-raf抑制剂组合的erk抑制剂;在某些实施例中,c-raf抑制剂是化合物a,并且erk抑制剂是化合物b。本文描述了用于使用此类方法的适合的剂量和给药方案。在一些实施例中,增殖性障碍是kras-突变型肿瘤,如本文所述的表达至少一种功能获得性kras突变的肿瘤,以及特别地,kras-突变型癌症,如nsclc(非小细胞肺癌)。包括的是具有braf突变(包括v600e和其他)的肿瘤,例如具有至少一种v600e或其他braf突变的nsclc,无论是典型的还是非典型的。用于本文披露的方法、治疗和组合的craf抑制剂是至少craf和任选还有braf的有效抑制剂。在一些实施例中,将craf抑制剂、或其药学上可接受的盐口服给予。在一些实施例中,craf抑制剂是化合物a、或其药学上可接受的盐。当将本文中的剂量描述为“约”指定量时,实际剂量可以从所述量变化高达10%,例如5%:这种“约”的使用承认,给定剂型中的精确量可能由于各种原因而与预期量略有不同,但不会实质上影响所给予化合物的体内作用。技术人员将理解,当本文引用治疗化合物的剂量(dose)或剂(dosage)时,该量是指游离形式的治疗化合物的量。单位剂量的craf抑制剂可以每日一次、或每日两次、或每日三次、或每日四次给予,其中实际给予剂量和给予时间由多个标准确定,如患者年龄、体重和性别;待治疗的癌症的程度和严重性;以及治疗医师的判断。在一个实施例中,制备化合物a用于口服给予,并且以100mg、150mg、200mg、250mg、300mg、或400mg的剂量口服给予(高达每日四次递送):基于动物中相应血浆水平的异速比例,预计每日一次或两次100mg的剂量可在人受试者中提供在人体中有效的血浆浓度,并且可给予200mg高达每日四次的剂量以在提供令人满意的治疗指标的同时获得更大的功效。在一些实施例中,将化合物a每日一次以100mg、200mg、250mg、300mg、或400mg的剂量给予。临床前模型的异速比例表明,作为单一药剂每日一次或每日两次、三次或四次分开给药的化合物a的300mg或更高的每日剂量在许多适应症(包括含有或表达kras突变的实体瘤)中应该提供治疗效果。在本发明的组合中,预期化合物a在这些受试者中的治疗剂量较低,因此组合治疗对这些受试者通常使用化合物a的100mg,200mg、250mg或300mg每日剂量。适当地,在本发明的组合和方法中,每日一次给予100mg、或200mg、或250mg、或300mg化合物a的剂量。在一个实施例中,制备化合物b用于通过口服递送给药,并且可以其盐酸盐形式使用。在一些实施例中,将化合物或其hcl盐简单地包封在药学上可接受的容器(如硬或软囊形片(gelcap))中用于口服给药。能以多种剂量生产囊形片,以便灵活给药;例如,可制备含有约5mg、约20mg、约50mg、或约100mg化合物b或其hcl盐的囊形片。在本发明的组合以及在本文描述的治疗用途中,将化合物a、或其药学上可接受的盐以约100mg、或约150mg、或约200mg、或约250mg的每日剂量,与化合物b、或其药学上可接受的盐(其能以约50mg、或约75mg、或约100mg或约150mg或约200mg的每日剂量给予)组合给予。例如,能以如下给予化合物a、或其药学上可接受的盐:每日约100mg化合物a的总剂量,以及每日约100mg化合物b、或其药学上可接受的盐的总剂量,该剂量优选地为每日给予一次。对其有需要的患者还可每日接受总剂量约200mg化合物a或其药学上可接受的盐,以及每日总剂量约100mg化合物b或其药学上可接受的盐,剂量优选每日给药一次。可以根据本文披露的方法一起使用化合物a和化合物b。取决于预期的给药剂量和频率,两种化合物能以任何顺序一起或分开给药,因为预期本发明的治疗可以持续如治疗医师认为合适的以及进一步使用本文所述的确定合适的剂量和频率的方法指导的2天、3天、4天、5天、6天、1周、2周、3周、4周或超过4周。在本文披露的方法和用途中,可以将化合物a和/或化合物b每日给予至少连续五日。在另一个方面,本发明的特征在于降低过度增殖性(例如癌症)细胞的活性(例如生长、存活、或生存力,或者全部)的方法。在另一方面,本发明提供了使用化合物b来治疗实体瘤的方法和组合物,将其与craf抑制剂分开、同时或顺序给予(或制备用于给予)。还提供了craf抑制剂,用于治疗在mapk途径中表达功能获得性突变的实体瘤,如kras-突变型nsclc(非小细胞肺癌)、braf-突变型nsclc(非小细胞肺癌)、kras-突变型和braf-突变型nsclc(非小细胞肺癌)、kras-突变型胰腺癌、kras-突变型结肠直肠癌(crc)和kras-突变型卵巢癌和brafv600-突变型黑素瘤,其中将craf抑制剂与erk抑制剂(如化合物b)分开、同时或顺序给予(或制备用于给予)。典型地,将化合物b口服给药,并与craf抑制剂分开、同时或顺序给予,craf抑制剂通常也口服给予。本文描述了用于这些方法和组合物的化合物a和化合物b的合适方法、给药途径、剂量和给药频率。在另一方面,本发明提供了用于治疗kras-突变型nsclc(非小细胞肺癌)和用于治疗braf突变型nsclc的erk1/2抑制剂,其中将erk1/2抑制剂与craf抑制剂分开、同时或顺序给予(制备用于给予)。还提供了craf抑制剂,用于治疗kras-突变型nsclc(非小细胞肺癌)以及用于治疗braf突变型nsclc,其中将craf抑制剂与erk1/2抑制剂分开、同时或顺序给予(或制备用于给予)。典型地,将erk1/2抑制剂口服给予,并且可以与craf抑制剂分开或顺序给予,该craf抑制剂也是口服给予。本文描述了craf抑制剂和erk1/2抑制剂给予的合适方法、途径、剂量和频率。在一些实施例中,craf抑制剂是化合物a;在一些实施例中,erk1/2抑制剂是化合物b。本文披露的组合可以以单一组合物一起给予或者以两种或更多种不同组合物(例如,如本文所述的组合物或剂型)分开给予。本文所述的药物组合,特别是本发明的药物组合,可以是自由组合产品,即两种或更多种活性成分(例如化合物a和化合物b,其作为两种或更多种不同的剂型同时、分开或顺序给予)的组合。治疗剂的给予可以是任何顺序。第一药剂和另外的药剂(例如第二、第三药剂)可以通过相同的给药途径或通过不同的给药途径给予。在另一方面,本发明特征在于包含化合物a和化合物b,任选还含有至少一种和任选地多于一种药学上可接受的赋形剂或载体的组合物。此类组合物用于治疗实体瘤,典型地是表达kras突变或raf突变的实体瘤,通常用于治疗nsclc,并特别是用于治疗具有展现至少一种kras突变(具体地是功能获得性突变,如本文所述的那些)的nsclc患者。非小细胞肺癌(nsclc)肺癌是影响全球男性和女性的常见癌症。非小细胞肺癌(nsclc)是最常见的肺癌类型(大致85%),其中近似70%的患者在诊断时呈现出疾病晚期(iiib期或iv期)。虽然肺癌通常与吸烟有关,但非吸烟者也会患肺癌,特别是nsclc:非吸烟者的诊断经常被延迟,因为与吸烟的关系很高,但10%-15%的肺癌患者从不吸烟。约30%的nsclc含有活化kras突变,并且这些突变与对egfrtki的抗性相关(paow,wangty,rielygj等人(2005)plosmed[公共科学图书馆医学];2(1):e17)。目前处于开发中的免疫疗法已开始为肺癌患者提供显著益处,包括常规治疗对其无效的患者。最近,派姆单抗(pembrolizumab,)和纳武单抗(nivolumab,)(pd-1/pd-l1相互作用的两种抑制剂)已被批准用于nsclc。然而,结果表明许多用单一药剂pd-1抑制剂治疗的患者不能从治疗中充分受益。直接抑制kras已被证明具有挑战性,并且kras-突变型nsclc仍然是癌症治疗的难以捉摸的靶标。迄今为止,没有经过批准的靶向治疗可用于患有kras-或braf突变型nsclc。已经在高达3%的nsclc中观察到braf突变,并且还已经将其描述为egfr突变阳性nsclc中的抗性机制(paikpk,arcilame,faram等人(2011)jclinoncol[临床肿瘤学杂志].5月20日;29(15):2046-51)。卵巢癌卵巢癌是最致命的妇科癌症,并且是由具有可变预后的不同组织学和分子亚型的集合组成的异质性疾病。上皮亚型包含90%的卵巢癌。上皮性卵巢癌中最常见的组织学亚型是浆液性癌,占上皮性卵巢癌的60%至70%。两层分级系统将浆液性癌分为低级浆液性(lgs)和高级浆液性(hgs),其具有不同的分子特性、免疫组织化学曲线、流行病学特征和临床表现。lgs癌占浆液性上皮性卵巢癌的10%,并且具有kras(高达40%)或braf突变(2%-6%)的卵巢癌主要是lgs癌。lgs癌不仅对一线药剂具有化学耐药性,而且在复发性疾病情况下也是如此。胰腺癌如本文所用,术语“胰腺癌”包括胰管腺癌(pdac)。pdac是胰腺癌的最常见的类型。kras-突变型癌症和braf-突变型nsclc本发明提供了包含(a)作为化合物a的craf抑制剂或其药学上可接受的盐、和(b)作为化合物b的erk抑制剂或其药学上可接受的盐的药物组合,用于在kras-突变型nsclc的治疗中使用。在另外的方面,本发明提供了包含(a)作为化合物a的craf抑制剂或其药学上可接受的盐、和(b)作为化合物b的erk抑制剂或其药学上可接受的盐的药物组合,用于在kras-突变型结肠直肠癌(crc)的治疗中使用。在另外的方面,本发明提供了包含(a)作为化合物a的craf抑制剂或其药学上可接受的盐、和(b)作为化合物b的erk抑制剂或其药学上可接受的盐的药物组合,用于在kras-突变型卵巢癌的治疗中使用。在另外的方面,本发明提供了包含(a)作为化合物a的craf抑制剂或其药学上可接受的盐、和(b)作为化合物b的erk抑制剂或其药学上可接受的盐的药物组合,用于在kras-突变型胰腺癌的治疗中使用。在另外的方面,本发明提供了包含(a)作为化合物a的craf抑制剂或其药学上可接受的盐、和(b)作为化合物b的erk抑制剂或其药学上可接受的盐的药物组合,用于在braf-突变型nsclc的治疗中使用。术语“braf-突变型”肿瘤或癌症包括展现突变的braf蛋白质的任何肿瘤。b-raf突变的实例包括但不限于v600e和v600k。大多数b-raf突变聚簇在两个区域:n叶的富含甘氨酸的p环和活化区段及侧翼区。已经在多种癌症中检测到v600e突变,所述v600e突变是由于在核苷酸1799处胸腺嘧啶被腺嘌呤取代。这导致在密码子600处缬氨酸(v)被谷氨酸(e)取代(现在称为v600e)。术语“kras突变型”肿瘤或癌症包括展现突变的kras蛋白(特别是功能获得性kras突变)的任何肿瘤;具体地是任何g12x、g13x、q61x或a146xkras突变型,其中x是除在该位置天然存在的氨基酸之外的任何氨基酸。例如,g12v突变意味着在密码子12处甘氨酸被缬氨酸取代。肿瘤中的kras突变的实例包括q61k、g12v、g12c和a146t。因此,kras-突变型nsclc、crc、卵巢癌和胰腺癌包括但不限于q61k、g12v、g12c和a146tnsclc,q61k、g12v、g12c和a146tcrc、卵巢癌或胰腺癌。例如,通过本文描述的组合疗法治疗的癌症包括krasq61k肺癌、krasg12d卵巢癌、krasg12d胰腺癌和krasg12r胰腺癌。术语“braf-突变型”肿瘤或癌症包括展现突变的braf蛋白质的任何肿瘤。b-raf突变的实例包括但不限于v600e和v600k。大多数b-raf突变聚簇在两个区域:n叶的富含甘氨酸的p环和活化区段及侧翼区。已经在多种癌症中检测到v600e突变,所述v600e突变是由于在核苷酸1799处胸腺嘧啶被腺嘌呤取代。这导致在密码子600处缬氨酸(v)被谷氨酸(e)取代(现在称为v600e)。被本文描述的药物组合治疗的癌症可以处于早期、中期或晚期状态。组合疗法的用途在一个方面,提供了治疗(例如减少、抑制或延迟进展中的一种或多种)受试者中增殖性疾病的方法,该增殖性疾病是具有一种或多种丝裂原活化蛋白激酶(mapk)改变的晚期实体瘤,如kras突变型肿瘤,并且具体地是kras突变型nsclc(非小细胞肺癌)。该方法包括向该受试者给予本文披露的组合(例如包含治疗有效量的erk1/2抑制剂和治疗有效量的化合物a或其药学上可接受的盐的组合)。本文所述的组合能以全身方式(例如口服、肠胃外、皮下、静脉内、直肠、肌肉内、腹膜内、鼻内、透皮、或通过吸入或腔内装置)、局部方式或通过应用于粘膜(如鼻子、喉咙和支气管)向该受试者给予。在一些实施例中,用于在这些组合和方法中使用的erk1/2抑制剂被口服给予。在一些实施例中,用于在这些组合和方法中使用的craf抑制剂被口服给予。当erk1/2抑制剂和craf抑制剂组合使用时,两者均可以口服给药,并且可以遵循由治疗医师确定的给药方案一起(同时)或以任何顺序分开给药;适合的剂量和给药方案在下文披露。另外的组合疗法在某些实施例中,将本文所述的方法和组合物与一种或多种其他癌症疗法模型如抗体分子、化学疗法、其他抗癌疗法药剂(例如靶向抗癌疗法、基因疗法、病毒疗法、rna疗法骨髓移植、纳米疗法或溶瘤细胞药物)、细胞毒性剂、基于免疫的疗法(例如细胞因子、免疫刺激剂或基于细胞的免疫疗法)、外科手术(例如乳房肿瘤切除术或乳房切除术)或放射程序、或前述中任何的组合中的组合给予。另外的疗法可以呈辅助或新辅助疗法的形式。在一些实施例中,另外的疗法是酶抑制剂(例如,小分子酶抑制剂)或转移抑制剂。可以与本发明的组合进行组合给予的示例性细胞毒性剂包括抗微管剂、拓扑异构酶抑制剂、抗代谢物、有丝分裂抑制剂、烷化剂、蒽环类药物、长春花生物碱、嵌入剂、能够干扰信号转导途径的试剂、促进细胞凋亡的试剂、蛋白酶体抑制剂和辐射(例如局部或全身辐射(例如γ辐射))。在其他实施例中,另外的疗法是手术或放射,或其组合。在其他实施例中,另外的疗法是靶向pi3k/akt/mtor途径、hsp90抑制剂或微管蛋白抑制剂中的一种或多种的疗法。可替代地,或与前述组合进行组合,本文所述的方法和组合物可与以下中的一种或多种进行组合给予:免疫调节剂(例如共刺激分子的活化剂或抑制分子的抑制剂,例如免疫检查点分子);疫苗,例如治疗性癌症疫苗;或其他形式的细胞免疫疗法。其他治疗剂、程序或模式的任何组合和顺序(例如,如本文所述)可以与本发明的治疗组合使用。本发明的组合物和组合可以在其他治疗方法之前、与其他治疗方法同时、在此类其他治疗的周期之间、或在障碍的缓解期间给予。本文公开了包含erk抑制剂和/或c-raf抑制剂的方法、组合和组合物,用于治疗癌症,具体地在mapk途径中表达至少一种功能获得性突变的实体瘤。选择术语在下面和整个申请中定义。如本文所用,冠词“一个/种(a和an)”是指一个/种或多于一个/种(例如,至少一个/种)该冠词的语法宾语。除非上下文另有明确说明,否则术语“或”在本文中用于表示术语“和/或”并且可与术语“和/或”互换使用。“约”和“大约”通常表示在给定测量的性质或精度的情况下测量的量的可接受的误差度。示例性误差度在给定值或值范围的20%内,典型地在10%内,并且更典型地,在5%内。特别地,当剂量被提及为“约”特定值时,其旨在包括在指定值±10%附近的范围。按照本领域的惯例,剂量是指游离形式的治疗剂的量。例如,当提及100mg化合物b的剂量,并且化合物b以其盐酸盐使用时,所用治疗剂的量相当于100mg游离形式的化合物b。“组合”或“与……组合”并不旨在暗示必须物理混合或同时给予疗法或治疗剂和/或配制这些治疗剂用于一起递送,尽管这些递送方法在本文所述的范围内。这些组合中的治疗剂可以与一种或多种其他另外的疗法或治疗剂同时、在其之前或之后给予。治疗剂或治疗方案可以以任何顺序给予。通常,每种药剂将以针对该药剂确定的剂量和/或计划表给予。还应理解,该组合中使用的另外的治疗剂可以以单一组合物一起给予或以不同组合物分开给予。通常,预期组合中使用的其他治疗剂的以不超过它们单独使用时的水平使用。在一些实施例中,组合中使用的水平将低于单一药剂疗法中使用的水平。如本文所用,术语“协同的”是指两种治疗剂(例如,化合物a的c-raf抑制剂化合物、和化合物b的erk1/2抑制剂化合物)的作用,其产生效果(例如,减缓增殖性疾病,特别地癌症、或其综合征的症状进展),该效果大于简单地添加每种药物自身给药的效果。例如,可以使用合适的方法,例如(lehar等人2009)中描述的那些计算协同效应。在实施例中,相对于单一药剂剂量水平,将另外的治疗剂(例如,craf抑制剂)以治疗剂量或低于治疗剂量的剂量给药。在某些实施例中,当第二治疗剂(例如,erk1/2抑制剂)与第一治疗剂组合使用或给予时,实现抑制(例如,生长抑制或肿瘤收缩)所需的第二治疗剂的浓度比单独给予第二治疗剂时低。在某些实施例中,当第一治疗剂与第二治疗剂组合给予时,实现抑制(例如,生长抑制)所需的第一治疗剂的浓度或剂量比单独给予第一治疗剂时低。在某些实施例中,在组合疗法中,实现抑制(例如,生长抑制)所需的第二治疗剂的浓度或剂量比作为单一疗法的第二治疗剂的治疗剂量低,例如低10%-20%、20%-30%、30%-40%、40%-50%、50%-60%、60%-70%、70%-80%、或80%-90%。在某些实施例中,在组合疗法中,实现抑制(例如,生长抑制)所需的第一治疗剂的浓度或剂量比作为单一疗法的第一治疗剂的治疗剂量低,例如低10%-20%、20%-30%、30%-40%、40%-50%、50%-60%、60%-70%、70%-80%、或80%-90%。术语“抑制”、“抑制剂”或“拮抗剂”包括给定分子或途径的某些参数(例如活性)的降低。例如,此术语包括将靶激酶(craf或erk1/2)的活性抑制5%、10%、20%、30%、40%或更多。因此,抑制可以是但不必是100%。术语“癌症”是指以异常细胞的不希望和不受控制的生长为特征的疾病。癌细胞可以局部或通过血流和淋巴系统扩散到身体的其他部位。如本文所用,术语“癌症”或“肿瘤”包括癌变前以及恶性癌症和肿瘤。如本文所用,术语“治疗(treat、treatment和treating)”是指由给予一种或多种疗法导致的障碍(例如增殖性障碍)的进展、严重性和/或持续时间的减少或缓解,或者障碍的一种或多种症状(优选地,一种或多种可辨别的症状)的缓解。在具体的实施例中,术语“治疗(treat、treatment和treating)”是指改善增殖性障碍的至少一种可测量的物理参数,如肿瘤的生长,这不一定是患者可辨别的。在其他实施例中,术语“治疗(treat、treatment和treating)”是指通过例如稳定可辨别的症状来物理地,或通过例如稳定物理参数来生理地,或通过两者,抑制增殖性障碍的进展。在其他实施例中,术语“治疗(treat、treatment和treating)”是指减少或稳定肿瘤大小或癌细胞计数。药物组合物和试剂盒调节剂量方案以提供最佳的希望反应(例如,治疗反应)。例如,如由治疗情况的紧急状态所指示的,可以给予单次推注,可以随着时间的过去给予若干个分次剂量,或可以按比例地减少或增加剂量。如本文所用的剂量单位形式是指适合作为用于待被治疗的受试者的单元剂量的物理上离散的单位;每个单位含有经计算与所要求的药物载体联合产生所希望的治疗效果的预定量的活性化合物。本发明剂量单位形式的规格是通过以下指定并且直接取决于以下:(a)活性化合物的独特特征和有待实现的具体治疗效果,以及(b)在混配这种活性化合物用于治疗个体的敏感性的领域中的固有限制。本发明的药物组合物可包括“治疗有效量”或“预防有效量”的本发明的化合物。“治疗有效量”是指以必要的剂量和在必要的时间段内有效实现所需治疗结果的量。治疗有效量将根据以下因素而变化:如个体的疾病状态、年龄、性别、和体重。治疗有效量还是其中craf和/或erk1/2的任何毒性或不利影响被治疗有益的效果胜过的一个量。“治疗有效剂量”优选以所希望的方式调节可测量的参数,例如相对于未治疗的受试者,肿瘤生长速率抑制至少约20%,更优选至少约40%、甚至更优选至少约60%,并且仍更优选至少约80%。可以在预测人肿瘤中的功效的动物模型系统中评估化合物理想地调节可测量参数(例如癌症)的能力,以帮助建立合适的剂量水平和方案。可替代地,组合物的此性质可以通过使用本领域技术人员已知的体外测定检查化合物调节不希望的参数的能力来评估。“预防有效量”是指以剂量计并且持续所需的时间段以实现所希望的预防结果的有效的量。典型地,因为预防的剂量是在疾病之前或早期在受试者体内使用的,所以这种预防有效量将小于治疗有效量。包含一种或多种本文所述化合物的试剂盒也在本发明的范围内。该试剂盒也可包括一种或多种其他要素:使用说明书;用于与所述一种或多种化合物使用的其他试剂;用于制备给予化合物的装置或其他材料,如混合容器;药学上可接受的载体;和用于对受试者给药的装置或其他材料,如注射器。本发明的组合具有治疗或保护功能或两者,并且可以在体内或离体使用。例如,可以将这些分子向在体外或离体培养的细胞或者人类受试者给予,以治疗、预防和/或诊断多种障碍,如本文所述的癌症。因此,在一个方面,本发明提供了通过将抗癌化合物与另一种抗癌化合物组合使用来增强该抗癌化合物功效的方法,特别是使用化合物a与化合物b一起以提供通过给予相似剂量任何一种化合物作为单一药剂不能安全实现的增强功效的方法。这些组合特别适用于治疗表达mapk途径中一种或多种功能获得性突变(特别是ras和/或raf基因的突变)的癌症。实例以下实例用于帮助理解本发明,但并不旨在且也不应解释为以任何方式限制其范围。实例1:n-(3-(2-(2-羟基乙氧基)-6-吗啉并吡啶-4-基)-4-甲基苯基)-2-(三氟甲基)异烟酰胺化合物a(化合物a)是具有以下结构的吗啉取代的联芳基化合物化合物a是披露在pct申请wo2014/151616中的实例1156。化合物a、化合物a的药学上可接受的盐和包含化合物a的药物组合物的制备也在该pct申请中披露,例如参见第739-741页。实例1a体外raf活性测定raf酶和催化失活的mek1蛋白质底物均使用常规方法在内部制备。将作为全长蛋白质(具有y340e和y341e活化突变)的crafcdna亚克隆进入用于sf9昆虫细胞表达的杆状病毒表达载体。将h14-3-3ζcdna亚克隆到用于sf9昆虫细胞表达的杆状病毒表达载体中。将共表达两种蛋白质的sf9细胞裂解并经受固定化镍层析,并用咪唑洗脱。使用第二柱(strepii结合柱)并用脱硫生物素洗脱。使用prescission酶除去蛋白质标签,并使用流穿步骤进一步纯化蛋白质以除去标签。c-raffl是指全长c-raf蛋白质。将具有失活的k97ratp结合位点突变的全长mek1用作raf底物。将mek1cdna用n-末端(his)6标签亚克隆到用于大肠杆菌表达的载体中。通过镍亲和层析然后进行阴离子交换从大肠杆菌裂解物中纯化mek1底物。将最终的mek1制剂生物素化(pierceez-linksulfo-nhs-lc-生物素)并浓缩。测定材料测定缓冲液:50mmtris、ph7.5、15mmmgcl2、0.01%牛血清白蛋白(bsa)、1mm二硫苏糖醇(dtt)终止缓冲液:60mm乙二胺四乙酸(edta)、0.01%20b-raf(v600e),活性的生物素化的mek,激酶死亡(kinasedead)α筛选检测试剂盒(alphascreendetectionkit)(从perkinelmertm可得,#6760617r)抗磷酸mek1/2(从细胞信号传导技术公司(cellsignallingtechnology,inc.)可得,#9121)384孔低体积测定板(white板)测定条件b-raf(v600e)大约4pmc-raf大约4nm生物素化的mek,激酶死亡大约10nm对于braf(v600e)10μmatp,并且对于craf1umatp在室温与化合物预孵育60分钟在室温反应时间1或3小时测定方案将raf和生物素化的mek,激酶死亡在测定缓冲液(50mmtris、ph7.5、15mmmgcl2、0.01%bsa和1mmdtt)中的2x最终浓度组合,并在测定板(greinerwhite384孔测定板#781207)中分配为5ml/孔,该测定板含有稀释在100%dmso中的0.25ml的40x的raf激酶抑制剂测试化合物。将板在室温孵育60分钟。通过添加5ml/孔在测定缓冲液中稀释的2xatp开始raf激酶活性反应。在3小时(b-raf(v600e))或1小时(c-raf)之后。将反应终止,并使用兔抗-p-mek(细胞信号传导公司(cellsignaling),#9121)抗体和α筛选igg(蛋白质a)检测试剂盒(perkinelmer#6760617r),通过向孔中添加10ml在终止/珠缓冲液(25mmedta、50mmtris、ph7.5、0.01%tween20)中的抗体(1:2000稀释)和检测珠(两种珠的1:2000稀释)的混合物来测量磷酸化产物。添加在黑暗条件下进行以保护检测珠免受光照。将盖子置于板的顶部并在室温下孵育1小时,然后在perkinelmerenvision仪器上读取发光。使用xlfit数据分析软件通过非线性回归计算对于每种化合物的50%抑制的浓度(ic50)。使用上文描述的测定,化合物a展现如以下报道的抑制功效。化合物a是b-raf和c-raf两者的ii类抑制剂。化合物b-rafic-50(μm)c-rafflic-50(μm)化合物a0.000730.00020实例1b化合物a在许多表达mapk途径中的突变的人癌症细胞系中表现出活性,如下表所示。注意,对于在braf或ras中至少有一种突变的细胞系,活性特别强。表1.化合物a对一组人癌症细胞系增殖的影响。实例1c为了研究化合物a在braf和/或mek抑制剂难以治疗的brafv600突变型黑素瘤细胞中的活性,评估了化合物a在来自表达mek1/2、nras的突变或braf的剪接变体的brafv600黑素瘤细胞系a375的机制模型中的抗增殖活性。已经在临床前研究和临床样品中证明了这些突变赋予braf和/或mek抑制剂抗性。与braf抑制剂维莫非尼(vemurafenib)和mek抑制剂司美替尼(selumetinib)的功效相比,化合物a在亲本a375细胞系及其表达各种突变等位基因的衍生物中的生长抑制作用总结如下。这些突变赋予对braf和mek抑制剂的抗性,导致ic50值增加超过50倍。相反,抗性模型仍对化合物a敏感,ic50仅增加2-3倍。这些数据支持化合物a在已经变得braf和/或mek抑制剂难以治愈的brafv600黑素瘤的患者中的使用。化合物a在对braf和mek抑制剂具有抗性的机制a375模型中的抗增殖作用实例1d用羟丙基甲基纤维素/羟丙甲纤维素、胶体二氧化硅、微晶纤维素、聚乙烯吡咯烷酮/聚维酮、和硬脂酸镁配制化合物a,并形成含有约50mg化合物a的片剂。将足以提供所希望剂量的许多片剂每日一次给予禁食的受试者。将受试者用100mg次/天、或200mg次/天的剂量治疗。在第一剂量的化合物a(第1周期第1天)后48小时、并且在多剂量(第1周期第15天)后多达24小时收集用于pk评估的系列血液样品。分别在给予单次100mg剂量和单次200mg剂量后4小时内实现447ng/ml和889ng/ml的最大血浆浓度(c最大)。分别在100mg和200mg剂量的化合物a后,第1天24小时剂量间隔(auctau)的平均血浆暴露剂量分别为5679hr.ng/ml和10019hr.ng/ml。患者的半衰期计算为约23-24小时。每日一次给药100mg导致化合物a在血浆中轻微积累,其中积累比为1.8。基于这些数据,建立了次/天的给药方案。随着此研究的进行,所有提供的数据都被认为是初步的。实例2:化合物a在kras突变型nsclc模型中的抗肿瘤活性h358模型:将携带肿瘤的scid米色雌性nci-h358小鼠(每组n=8)在肿瘤细胞接种后14天随机分成3组,其中平均肿瘤体积范围为259.44-262.47mm3。在治疗过程中,向动物给予口服剂量30mg/kg或200mg/kg的媒介物或化合物a,连续14天,给药体积为10ml/kg动物体重。通过数字卡尺每周测量3次肿瘤体积,并在整个治疗过程记录所有动物的体重。calu6模型:将携带肿瘤的calu6雌性裸小鼠(每组n=6)在肿瘤植入后第17天随机分入治疗组,此时平均肿瘤体积为180mm3。用化合物a治疗在第17天开始并持续16天。给药体积为10ml/kg。在随机化时收集肿瘤体积,并且在之后研究持续时间内每周收集两次。h727模型:将携带肿瘤的nci-h358雌性裸小鼠(每组n=8)随机分成2组,其中平均肿瘤体积范围为275.74mm3。在治疗过程中,向动物给予口服剂量100mg/kg的媒介物或化合物a,连续14天,给药体积为10ml/kg动物体重。通过数字卡尺每周测量3次肿瘤体积,并在整个治疗过程记录所有动物的体重。如图1a、1b和1c所示,化合物a在krasmtnsclc模型中显示单一药剂活性。在基于细胞的测定中,化合物a在细胞系中证明了抗增殖活性,所述细胞系含有活化mapk信号传导的多种突变。例如,化合物a抑制非小细胞肺癌细胞系calu-6(krasq61k)、结肠直肠细胞系hct116(krasg13d)的增殖,其中ic50值范围为0.2μm-1.2μm。在体内,用化合物a治疗在若干种人kras突变型模型中产生肿瘤消退,这些kras突变型模型包括nsclc衍生的calu-6(krasq61k)和nci-h358(krasg12c)异种移植。在所有情况下,如通过缺乏显著的体重减轻判断的,抗肿瘤作用是剂量依赖性的并且耐受性良好。当植入裸小鼠和裸大鼠时,calu-6模型对化合物a敏感,其中在小鼠中在剂量为100mg/kg、200mg/kg和300mg/kg(每日一次(qd))时观察到消退并且在大鼠中在75mg/kg和150mg/kg(qd)时观察到消退。在小鼠和大鼠中分别在30mg/kgqd和35mg/kgqd时观察到该模型中的肿瘤停滞。在小鼠第二人nsclc模型nci-h358中200mg/kgqd剂量和在小鼠人卵巢hey-a8异种移植物中低至30mg/kgqd的剂量也实现了消退。此外,来自calu-6异种移植物中的剂量分级功效研究的数据证明,在不同给药水平下,每日一次(qd)给予化合物a和每日两次(bid)分次给予化合物a显示出相似水平的抗肿瘤活性。这些结果支持在临床中探索qd或bid剂量方案。总体而言,在耐受良好的剂量下观察到的化合物a的体外和体内mapk-途径抑制和抗增殖活性表明化合物a在具有mapk途径中活化病变的肿瘤的患者中可以具有抗肿瘤活性,并且具体地可以因此可用作单一药剂或与第二药剂(如影响mapk途径的不同步骤的抑制剂)组合用于治疗具有kras突变的nsclc患者。已经显示化合物a作为单一药剂具有针对在mapk途径中表达功能获得性突变的各种其他癌症的活性,例如,在ras或raf中,包括卵巢癌、胰腺癌和黑素瘤,并且在与erk抑制剂(如化合物b)组合使用时,已经显示在模型系统中对这些病症更有效。实例3:化合物b是erk1/2的抑制剂。披露了化合物,并且其制备描述于公开的pct专利申请wo2015/066188中。在一些实施例中,将此化合物以其盐酸盐使用。在3天的增殖测定中,化合物b显示细胞生长的强烈抑制,其中在具有kras突变的细胞系(包括肺癌细胞系calu6(krasq61k)、卵巢癌细胞系heya8(krasg12d)、胰腺癌细胞系aspc-1(krasg12d)和psn1(krasg12r))的亚组中ic50值小于1μm。实例4:化合物a和化合物b的组合对于kras突变型细胞系的影响在一组来自nsclc(4)、结肠直肠癌(crc)(4)和胰腺导管腺癌(pdac)(6)的十四(14)个kras突变细胞系中体外评估化合物a和化合物b组合对增殖的影响。(ctg)荧光细胞生存力测定试剂盒(普洛麦格公司(promega),麦迪逊,wi,usa)测量了裂解细胞后孔中存在的atp的量。裂解后释放的atp在酶促反应中测量,其包括荧光素酶及其底物荧光素。发射的光量与atp的量成比例,atp反过来与孔中活细胞的数量成比例。使用此测定来确定药物治疗后活细胞的比例。用于ctg测定(包括14种细胞系、培养基和细胞密度)的试剂描述于下表中。根据下表中的信息,以50ul/孔将细胞一式两份接种于在白色384孔组织培养板(#3707,科宁(corning),ny,usa)中。第二天,将化合物在dmso中在复合板(#788876,葛莱娜公司(greiner),monroe,nc,usa)中稀释,产生7点1:3稀释系列(所需最终浓度的1000倍)。使用声学分配器(ats100,edc生物系统公司(edcbiosystems),加利福尼亚,usa)将化合物分配至细胞板以实现1:1000稀释。例如,复合板的一个孔含有10mm化合物a,并且将50nl化合物转移到50ul细胞中,对于该给定的孔中的化合物实现10um的最终浓度。2.3节概述了使用ats100在我们的测定板中创建的组合网格的布局和浓度。随后将测定板返回到37℃的潮湿co2培养箱中。在5天的化合物孵育后,向每个孔中添加25ul/孔的ctg生存力试剂(总体积为75ul),并在室温下孵育15分钟后,使用0.1秒超灵敏发光方案在酶标仪上读板(envision,珀金埃尔默公司(perkinelmer),hopkington,ma,usa)。将数据归一化为对于给定细胞系的未处理孔(仅用dmso处理)的平均值。然后从1中减去值并乘以100以表示%抑制。然后通过专有软件(helios)的四参数逻辑(4pl)回归曲线拟合模型将归一化数据拟合到曲线。报道了单一药剂曲线的ic50(半数最大抑制浓度),其中化合物抑制50%细胞生长。表:测定试剂和材料表:细胞系培养基和接种密度在所有情况下,使用浓度范围为0.014至10um的化合物a、浓度范围为0.011至8.0um的化合物b,在棋盘格式化矩阵中评估组合。使用两种协同作用测量来确定组合在特定细胞系中是否具有协同作用;协同评分和组合指数(ci)在50%或75%抑制水平(lehar等人2009)。在测试的7/14细胞系中,化合物a和化合物b的组合中度地至强度地协同。对于每种细胞系的这些值得总结显示在以下表中。表:化合物ax化合物b协同作用评分和ci50值得总结在以下表中提供了对评分/值进行解释的一般准则。表:解释组合活性在测试的细胞系组中,在源自nsclc和pdac谱系的模型中显著地观察到协同作用,其中calu-6(krasq61k)和hupt4(krasg12v)模型分别对两个谱系响应最强。实例5:在疾病的异种移植模型中化合物a与化合物b组合。通过皮下注射在50%matrigeltm中1000万个细胞到每只小鼠的侧腹,在无胸腺雌性小鼠中建立calu-6nsclc肿瘤。当肿瘤达到大约250mm3时,根据肿瘤体积将小鼠随机分入治疗组(n=7)。每日一次(qd)或每隔一日(q2d)以所示剂量水平口服给予测试剂。a-b)治疗组的肿瘤体积与随机化后的天数。c)与最初相比的体重平均百分比变化。这些治疗的数据显示在图1a、1b和1c中。与人nsclc异种移植物calu-6中的任一单一药剂相比,化合物a和化合物b的组合疗法导致肿瘤应答的深度和持久性增加。17天给药后,分别地,以30mg/kgqd给药的化合物a实现26%t/c,而以75mg/kgq2d或50mg/kgqd给药的化合物b实现4%t/c和22%消退。17天给药后,30mg/kgqd给药的化合物a与在50mg/kgqd或75mg/kgq2d的化合物b组合分别实现66%和51%消退。除了增加的响应深度之外,化合物a和化合物b的组合还导致响应的持久性增加。虽然给予单一药剂化合物a和化合物b的小鼠肿瘤在治疗下进展,但将化合物a和化合物b组合在一起(无论化合物b的剂量如何)在给药后42天维持肿瘤消退。总之,这些数据表明,用化合物a和化合物b的组合治疗可以在具有由于mapk途径中的功能获得性突变而活化的mapk途径的患者中实现更大和更持久的响应。此外,这些结果支持当与化合物a组合时化合物b间歇给药的潜在探索。通过体重没有减轻判断,所测试的组合具有良好的耐受性。实例6:在具有mapk途径改变的晚期实体瘤的成人患者中单独或与化合物b一起的化合物a的i期剂量发现研究。化合物a作为单一药剂根据临床前安全性、耐受性数据、临床前研究中获得的pk/pd数据以及探索性人有效剂量范围预测,本研究中化合物a单一药剂的推荐起始剂量和方案为口服100mgqd。可以使用100mg、200mg、250mg、300mg、或400mg的起始剂量;初步数据表明,每日一次250mg的起始剂量可以对实体瘤有效。为了最大的给药灵活性,可以将化合物a制备成50mg和/或100mg片剂用于口服给药。用于临床使用化合物a的所提出的制剂包括与一种或多种选自以下的赋形剂的组合的化合物a:羟丙基甲基纤维素(羟丙甲纤维素)、胶体二氧化硅、微晶纤维素、聚乙烯吡咯烷酮(聚维酮)和硬脂酸镁,并且能以片剂的形式制备用于口服给药,适当地含有50mg或100mg化合物a。剂量递增的暂定剂量可以在下表中找到。表化合物a的暂定剂量水平在剂量扩增部分中,化合物a单一药剂组中的患者用化合物a以基于剂量递增数据选择的推荐剂量和方案进行治疗。在试验中包括的所有适应症中,预期该剂量对成人患者是安全的和耐受的。该首次人体试验的临床方案是化合物a的每日连续一次给药方案。已经证明qd方案在临床前研究中是有效和耐受的。在calu6异种移植物中,使用qd或分次的bid方案实现了相似的功效水平,表明功效与总体暴露有关。预测的人pk和预测的半衰期(约9h)也表明qd给药可以实现有效的暴露。另外,可以通过监测指示map激酶途径抑制的生物标记来确定有效剂量。特别地,dusp6(双特异性磷酸酶6)是此途径的已知生物标记,并且已经显示dusp6的体内水平在接受化合物a的剂量的受试者中下降,这与化合物a的有效血浆水平相关。因此,dusp6可以用作用化合物a治疗的受试者的药效学生物标记,无论是作为单一药剂还是作为与另外的种治疗剂的组合。化合物a与化合物b组合化合物b可以以其盐酸盐使用。出于测试目的,可以口服给予,与一种或多种赋形剂一起配制,或者作为单独的化合物,包含在药学上可接受的胶囊例如软或硬囊形片中、或者与方便的培养基混合以通过填喂法给药。化合物a与化合物b组合的剂量递增开始于将化合物a鉴定为单一药剂的给药方案:化合物a的起始剂量低于单一药剂剂量。因此,此剂量的选择应当最小化对潜在毒性药物水平的暴露,同时限制可能接受剂量太低而不能提供良好功效的患者数量。化合物a的方案与针对单一药剂化合物a所选择的方案相同。如果在单一药剂扩增部分期间将探索化合物a单一药剂的两种方案,则将基于包括安全性和暴露的所有可用数据选择一种优选方案用于该组合。可以基于新出现的数据来决定在后期阶段转换联用组中的化合物a剂量方案。将化合物b以预期的剂量水平给予,以提供足以基于临床前模型中的活性提供有效肿瘤抑制的暴露。基于临床前测试,功效似乎由高于有效血浆水平的时间驱动,因此在一个实施例中可通过监测化合物b的血浆水平来确定剂量。预计所希望的针对功效的最低血浆水平为约600nm,或约350ng/ml;因此,剂量和给药方案(频率)可以通过将化合物b的血浆浓度维持在高于此最低血浆水平持续至少一天、或至少一周,或如1、2、3、或4周的治疗周期的目标(由治疗医师判断适当的)来指导。此试验的临床方案是化合物a和化合物b的连续每日一次给药方案。从临床试验获得的初步结果进一步证实了这一点。根据实体瘤反应评估标准(recist),显示用1200mgqd的化合物a治疗的患非小细胞肺癌(nsclc)的受试者导致-35%的部分反应。在剂量扩增部分中,基于剂量递增数据,联用组中的患者以推荐剂量和药物组合方案进行治疗。为了提供合适的剂量灵活性,化合物b能以含有各种量的化合物b(任选地作为其盐酸盐)的囊形片的形式制备,例如每个囊形片约5mg、20mg、50mg、或100mg的剂量。组合组中的患者可以用约100mg、或约150mg、或约200mg、或约250mg的每日剂量的化合物a以及约50mg、或约75mg、或约100mg的每日剂量的化合物b治疗。例如,可以每日给予患者总剂量约100mg化合物a,以及每日总剂量约100mg化合物b,剂量优选每日给药一次。患者还可接受每日总剂量约200mg化合物a,以及每日总剂量约100mg化合物b,剂量优选每日给药一次。接受组合疗法的患者包括nsclc患者,例如成年患者,患有晚期或转移性kras-突变型或braf突变型(例如,brafv600e-突变型)nsclc。按照标准护理这些患者可以已经进展,或者对于其没有有效的标准治疗方法。可以评估试验的功效,根据recist版本1.1和总体存活(os)测量总响应率(orr)、疾病控制率(dcr)、响应持续时间(dor)、无进展存活(pfs)。等效物虽然已经讨论了本发明的特定实施例,但上述说明是说明性而非限制性的。在综述本说明书和以下权利要求书之后,本发明的许多修改对于本领域的技术人员将是显而易见的。应当通过参考权利要求书及其等同物的全范围以及说明书连同此类变化来确定本发明的全范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1