亲水性药物的油相液晶凝胶前体制剂及其制备方法与流程
本发明属于药物前体制剂领域,具体涉及亲水性药物的油相液晶凝胶前体制剂及其制备方法。
背景技术:
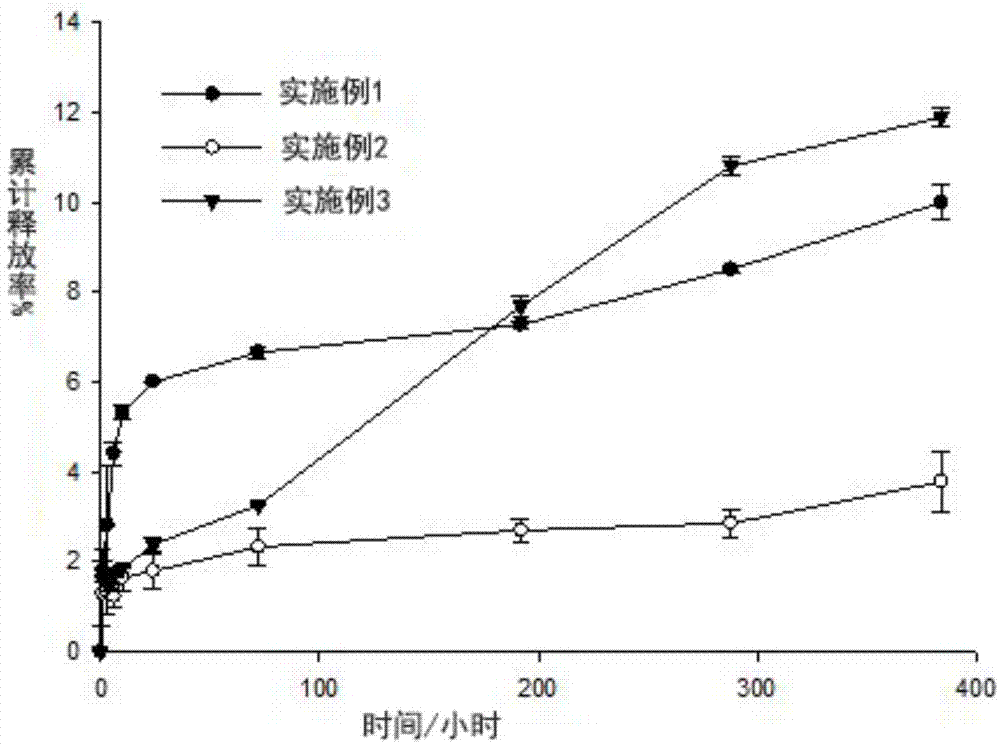
药物通过不同的药方式可以分为口服制剂、注射制剂、经鼻制剂、透皮制剂等。不同的给药方式需要考虑不同的因素。比如对于分子量大,半衰期短、脂溶性差的多肽蛋白质类药物,口服制剂经过胃酸和胃蛋白酶容易产生降解失活;注射剂需要考虑载体的生物相容性等;透皮制剂要考察个体及作用部位的差异对药物的吸收影响等等。另外如何提高药物的存储稳定性、减少给药频率、提高药物的生物利用度、降低药物副作用,在药物的开发和研究中也极其重要。许多药物的活性剂在体内浓度维持在一定范围内才会产生有益作用,过高或者过低都达不到期望的效果,药物浓度过高还会导致毒副作用。另外对于一些半衰期较短的药物长期高频给药会使病人的顺应性较差。针对上述问题,有必要开发具有较好稳定性的缓释剂,可以有效地控制药物释放速度,降低副反应,减少服药次数,提高病人顺应性。溶致液晶凝胶主要是由一种或多种双亲化合物和溶剂形成的有序的体系。由于其内部有一个一个晶格组成,晶格间的有序排列以及两亲分子间的强烈相互作用力使其外观上是凝胶状。溶致液晶由于其自身结构的特点吸引了众多科学家的关注,并逐渐用于多种药物的载体,在体内可以长效缓释各种极性药物,使药物在体内长时间维持有效浓度,达到提高其生物活性、减少给药次数的目的。现有的凝胶制剂技术中已有采用磷脂和甘油酯作为药物载体,由于不同药物的极性不同,为了解决有些药物在体系中溶解性差的问题,一般会采用加入大量溶剂及表面活性剂的方法,将药物溶解在磷脂或甘油酯或者溶剂中后,可以将各组分混合后制成注射剂,也再加入水制成液晶纳米粒。注射剂进人体后与人体内水产生反应,形成凝胶,起到药物缓释的作用。液晶纳米粒可以作为口服制剂或鼻腔给药等,存储稳定性好,且具有良好的缓释作用。如专利cn108272747a公开了一种亲水性药物的油相液晶凝胶前体制剂及其制备方法,中国专利cn108309931a公开了一种治疗滑膜炎的制剂及其制备方法。中国专利cn108283621a公开了一种鼻腔纳米制剂糠酸莫米松液晶凝胶纳米粒及其制备方法。然而对于亲水性的药物,尤其是亲水性大分子药物,如蛋白质igg、pd-1、pd-l1、胰岛素,制成水相制剂稳定性较差,容易水解;而目前市场上的亲水性药物油相制剂不能形成凝胶,普遍存在缓释效果不佳的问题;且存放稳定性较差。亲水性药物在油相中难以溶解,需要加入水相才能溶解,因而难以制成均匀油相的液晶凝胶前体制剂。因此急需要开发一种新的亲水性药物的油相制剂制备方法,使得亲水性药物可以制成均匀的油相制剂,既具有缓释作用还可以具有良好的稳定性。技术实现要素:本发明旨在至少解决现有技术问题之一,本发明提供一种具有长效缓释作用的亲水性药物的油相液晶凝胶前体制剂及其制备方法。本发明的技术方案为:亲水性药物的油相液晶凝胶前体制剂的制备方法,包括以下步骤:(s1)磷脂分散体的制备:称取磷脂和蒸馏水以1:3~5的质量比加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取亲水药物加水完全溶解,制成1~20mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体与步骤(s2)中制得的药物溶液以1:3~2:3的体积比混匀得到水相;(s4)油相的制备:在甘油酯中加入0.01%~0.05wt%的表面活性剂,混匀得到油相;(s5)油相水相混匀:称取适量的步骤(s4)制得的油相加入步骤(s3)制得的水相中使混合物中磷脂和甘油酯的质量比为32:68~70:30,涡旋混匀后,液氮立即冻干,得到预冻好的样品;(s6)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s7)制备混合油相:分别按质量比32:68~70:30称量好磷脂和甘油酯混合,再加入5~20wt%的助溶剂,放在35~40℃的摇床上振摇至完全溶解,得混合油相;(s8)制备药物前体制剂:取适量步骤(s7)制得的混合油相加入步骤(s6)制得的冻干样品中,放在35~40℃的摇床上振摇至完全溶解,得到澄清透明的液晶凝胶前体制剂,密封、备用。本发明的方案可以用于亲水性小分子药物如维生素,也可以用于亲水性大分子如抗体蛋白和胰岛素等,也可用于小分子和大分子亲水性药物的共溶。本方案采用磷脂分散体和药物水溶液混合后再与油相混合,再冻干除水后,加入磷脂和甘油酯的混合油相,即可以解决亲水性分子不溶于甘油酯的问题,提高亲水性药物的溶解度,并且制得的药物前体制剂具有油磷脂和甘油酯的多层保护,存放稳定性好。药物前体制剂可以作为注射剂,进入人体遇水形成凝胶,可以达到良好的缓释的效果,延长药物的作用时间,减少药物用药次数,且提高其生物利用度。所述亲水性药物为不溶于甘油酯的大分子药物时,优选为蛋白质igg、pd-1、pd-l1或胰岛素中的一种或多种,制得的药物前体制剂中亲水药物浓度为1~20mg/ml。本发明尤其适合本方案中的亲水性大分子药物,可以显著改善其在存放稳定性,提高药物存放活性和体内活性,并通过缓释延长给药时间,提高病人的适应性。优选地,所述的助溶剂为乙醇、中链脂肪酸、长链脂肪酸、花生油、大豆油、玉米油、蓖麻油、芝麻油中至少一种。进一步优选地,所述助溶剂为乙醇。优选的助溶剂生物相容性好,可以提高药物的溶解度,且粘度低,适用于注射,不会引起机体的炎症反应。优选地,表面活性剂为非离子型表面活性剂,更优选为吐温80。优选地,所述的甘油酯为二油酸甘油酯、三油酸甘油酯、硬脂酸甘油酯、棕榈酸甘油酯中至少一种。进一步优选地,所述的甘油酯为二油酸甘油酯。优选的甘油酯成本更低,生物相容性好,且配合磷脂可以更快与水反应形成稳定性更好的液晶凝胶。优选地,所述磷脂为大豆磷脂、磷脂酰乙醇胺、磷脂酰丝氨酸、磷脂酰甘油中的至少一种。所述方案优选了成本低、生物相容性好、双亲性好的磷脂,使体系可快速与水反应形成液晶凝胶,且药物的生物利用度较高。优选地,所述磷脂中由80~90wt%的大豆磷脂、5~10wt%的磷脂酰乙醇胺、5~10wt%的磷脂酰丝氨酸组成。上述混合磷脂提高混合油相的稳定性,使亲水药物的稳定性更好。综上所述,本发明的有益效果为:亲水性药物溶于混合油相形成油相制剂,存放稳定性好;可以用于注射,且注射到体内遇水形成凝胶,凝胶制剂的缓释作用可以使药物较长时间维持有效浓度,减少用药次数;原料生物相容性好,不会引起机体炎症反应。附图说明图1为实施例1-3制得的亲水性药物的油相液晶凝胶前体制剂中亲水性药物的累积释放率图;图2不同比例的spc/gdo样品的流变学性质;图3不同比例spc/gdo的凝胶在不同脂肪酶含量作用下的降解情况。具体实施方式为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。除非特别说明,本发明采用的试剂、方法和设备为本
技术领域:
常规试剂、方法和设备。实施例1本实施例亲水性药物的油相液晶凝胶前体制剂,由以下步骤的制备方法制得:(s1)磷脂分散体的制备:称取磷脂酰甘油1g和蒸馏水4g加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散20min,50%(650w),1s/3s,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取蛋白质pd-15mg加1.5ml水完全溶解,制成3.33mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体140ul与步骤(s2)中制得的药物溶液300ul混匀得到水相;(s4)油相的制备:在2.7g二油酸甘油酯中加入0.3mg的吐温80,混匀得到油相;(s5)油相水相混匀:称取65mg步骤(s4)制得的油相加入步骤(s3)制得的水相中,涡旋混匀后,液氮立即冻干,得到预冻好的样品;(s6)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s7)制备混合油相:分别称175mg磷脂酰甘油和325mg二油酸甘油酯混合,再加入25mg的中链脂肪酸,放在37℃的摇床上振摇至完全溶解,得混合油相;(s8)制备药物前体制剂:取400ul步骤(s7)制得的混合油相加入步骤(s6)制得的冻干样品中,放在37℃的摇床上振摇至完全溶解,得到澄清透明的药物前体制剂,密封、备用。所得前体制剂的蛋白质pd-1浓度为2mg/ml。本实施例制得的亲水性药物前体制剂4℃放置6个月、9个月、12个月后,离心处理,均无现象,说明制剂存放稳定性好。本发明制备的亲水性药物前体制剂进行亲水性药物释放考察实验方法:称取适量的亲水性药物的油相液晶凝胶前体制剂于洁净干燥的ep管,然后加入一定体积的ph7.4磷酸盐缓冲液后,密封,放入恒温摇床中进行振荡释放,恒温摇床参数设置:温度37℃,转速100rpm。分别于不同时间点取样,然后向释放体系中补加相同体积的释放介质。取出的释放介质放置-20℃备用,经hplc检测释放介质中亲水性药物的含量。本实施例亲水性药物的油相液晶凝胶前体制剂中蛋白质pd-116天的累积释放率约为10%,具有良好的缓释效果,可以大大延长用药周期,减少用药次数。实验结果如图1所示。油相中磷脂与油脂的比例不同,缓释效果也不同。实施例2本实施例亲水性药物的油相液晶凝胶前体制剂,由以下步骤的制备方法制得:(s1)磷脂分散体的制备:称取大豆磷脂1g和蒸馏水4g加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散20min,50%(650w),1s/3s,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取蛋白质igg5mg加1.5ml水完全溶解,制成3.33mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体200ul与步骤(s2)中制得的药物溶液300ul混匀得到水相;(s4)油相的制备:在2.7g二油酸甘油酯中加入0.3mg的吐温80,混匀得到油相;(s5)油相水相混匀:称取50mg步骤(s4)制得的油相加入步骤(s3)制得的水相中,涡旋混匀后,液氮立即冻干,得到预冻好的样品;(s6)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s7)制备混合油相:分别称250mg大豆磷脂和250mg二油酸甘油酯混合,再加入50mg的乙醇,放在37℃的摇床上振摇至完全溶解,混合油相;(s8)制备药物前体制剂:取400ul步骤(s7)制得的混合油相加入步骤(s6)制得的冻干样品中,放在37℃的摇床上振摇至完全溶解,得到澄清透明的药物前体制剂,密封、备用。所得制剂的蛋白质igg浓度为2mg/ml。本实施例制得的亲水性药物前体制剂4℃放置6个月、9个月、12个月后,离心处理,均无分层现象,说明制剂存放稳定性好。本实施例中蛋白质igg16天的累积释放率约为3.8%,具有良好的缓释效果,可以大大延长用药周期,减少用药次数。实施例3本实施例亲水性药物的油相液晶凝胶前体制剂,由以下步骤的制备方法制得:(s1)磷脂分散体的制备:称取大豆磷脂1g和磷脂酰乙醇胺0.3g和蒸馏水4g加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散20min,50%(650w),1s/3s,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取胰岛素20mg加1ml水完全溶解,制成20mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体210ul与步骤(s2)中制得的药物溶液300ul混匀得到水相;(s4)油相的制备:在2.7g棕榈酸甘油酯中加入1mg的吐温80,混匀得到油相;(s5)油相水相混匀:称取30mg步骤(s4)制得的油相加入步骤(s3)制得的水相中,涡旋混匀后,液氮立即冻干,得到预冻好的样品;(s6)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s7)制备混合油相:分别称250mg大豆磷脂和250mg棕榈酸甘油酯混合,再加入50mg的乙醇,放放在37℃的摇床上振摇至完全溶解,混合油相;(s8)制备药物前体制剂:取400ul步骤(s7)制得的混合油相加入步骤(s6)制得的冻干样品中,放在37℃的摇床上振摇至完全溶解,得到澄清透明的药物前体制剂,密封、备用。所得制剂的胰岛素浓度为10mg/ml。本实施例制得的亲水性药物前体制剂4℃放置6个月、9个月、12个月后,离心处理,均无分层现象,说明制剂存放稳定性好。本实施例中胰岛素16天的累积释放率约为11.5%,具有良好的缓释效果,可以大大延长用药周期,减少用药次数。实施例4相比实施例2,本实施例中磷脂由80wt%的大豆磷脂、10wt%的磷脂酰乙醇胺、10wt%的磷脂酰丝氨酸组成;其它制备方法和使用原料均与实施例2相同。对比例1相比实施例2,本对比例亲水性药物的油相液晶凝胶前体制剂的制备方法中:步骤(s1)到(s6)与实施例2相同,步骤(s7)改为取400ul二油酸甘油酯加入步骤(s6)制得的冻干样品中,放在37℃的摇床上振摇至完全溶解,得到药物前体制剂,得到澄清的药物前体制剂,密封、备用。所得制剂的蛋白质igg浓度为2mg/ml。本对比例制得的亲水性药物前体制剂4℃放置6个月、9个月后无分层,12个月后出现分层,9个月后离心处理也分层现象,说明制剂存放稳定性较实施例差。对比例2本对比例提供亲水性药物的油相液晶凝胶前体制剂的制备方法如下:(s1)磷脂分散体的制备:称取大豆磷脂1g和蒸馏水4g加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散20min,50%(650w),1s/3s,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取蛋白质igg5mg加1.5ml水完全溶解,制成3.33mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体200ul与步骤(s2)中制得的药物溶液300ul混匀得到水相;液氮立即冻干,得到预冻好的样品;(s4)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s5)制备混合油相:分别称250mg大豆磷脂和250mg二油酸甘油酯混合,再加入50mg的乙醇,放在45℃的烘箱完全溶解混匀,混合油相;(s6)制备药物前体制剂:取400ul步骤(s5)制得的混合油相加入步骤(s4)制得的冻干样品中,放在37℃的摇床上振摇至完全溶解,得到药物前体制剂,密封、备用。所得制剂的蛋白质igg浓度为2mg/ml。本对比例制得的亲水性药物前体制剂4℃放置6个月、9个月、12个月后出现无分层,12个月后离心处理出现分层现象,说明制剂存放稳定性较实施例差。对比例3本对比例提供亲水性药物的油相液晶凝胶前体制剂的制备方法如下:(s1)磷脂分散体的制备:称取大豆磷脂1g和蒸馏水4g加入瓶中,封口,放在摇床上分散成粗分散体;将粗分散体冰浴条件下探头超声分散20min,50%(650w),1s/3s,形成乳白色磷脂分散体;(s2)亲水性药物溶液的制备:称取蛋白质igg5mg加1.5ml水完全溶解,制成3.33mg/ml的药物溶液备用;(s3))水相的制备:取步骤(s1)中制得的磷脂分散体200ul与步骤(s2)中制得的药物溶液300ul混匀得到水相;液氮立即冻干,得到预冻好的样品;(s4)制备冻干样品:将预冻好的样品冻干除去水相介质,制得冻干样品;(s5)油相的制备:在2.7g二油酸甘油酯中加入0.3mg的吐温80,混匀得到油相;(s6)制备药物前体制剂:取400ul步骤(s5)制得的油相加入步骤(s4)制得的冻干样品中,混匀,得到药物前体制剂,密封、备用。所得制剂的蛋白质igg浓度为2mg/ml。本对比例制得的亲水性药物前体制剂4℃放置6个月、9个月后无分层,12个月后出现分层,9个月后离心处理也分层现象,说明制剂存放稳定性较实施例差。对比例4一种亲水性药物的制备方法,包括以下步骤:将20mg胰岛素、450mg磷脂、450mg二油酸甘油酯和90mg乙醇和30ug吐温80混合溶解,即得胰岛素液晶凝胶前体制剂。本对比例制得的胰岛素液晶凝胶前体制剂在4℃放置6个月后出现分层现象,说明本对比例制剂存放的稳定性相比各组实施例制剂要差。对比例5本对比例的药物前体制剂为直接加水溶解蛋白质igg制成浓度为2mg/ml的溶液。对比例6本对比例的药物前体制剂为直接加水溶解胰岛素,制成浓度为10mg/ml的溶液。将对比例和实施例2和实施例4中igg液晶凝胶前体制剂释放度实验释放的igg蛋白用乙醇沉淀,从对比例、实施例2和4中取等量igg作为样品,采用上海通蔚生物科技有限公司的免疫球蛋白g(igg)elisa试剂盒进行酶联免疫吸附试验,具体步骤同试剂盒操作说明书,实验结果见表1。表1不同组别igg酶联免疫吸附试验结果从表1可以看出,对于igg液晶凝胶前体制剂,本发明实施例2和4制剂释放的具有生物活性的igg蛋白含量较对比例高,说明本发明制剂释放的的igg蛋白结构完整、活性稳定,很可能是由于本发明方法中磷脂和甘油酯对igg蛋白的形成多层保护作用使得igg蛋白稳定性好,在冻干和复溶过程中减少了igg蛋白的降解或变性而造成的损失。将实施例3和对比例中胰岛素液晶凝胶前体制剂释放度实验释放的胰岛素用乙醇沉淀,取对比例和实施例3中等质量的胰岛素作为样品,按照《中华人民共和国药典》(2015年版四部)通则1211胰岛素生物测定法进行测定,实验结果见表2。表2不同组别胰岛素生物测定结果组别实施例3对比例4对比例6血糖值(mmol/l)6.3±1.07.1±1.27.6±1.1从表2可以看出,对于胰岛素液晶凝胶前体制剂,本发明实施例3制剂的降血糖效果较对比例4和6更优,说明本发明制剂的胰岛素生物活性更高,很可能是由于本发明方法中磷脂和甘油酯具有对胰岛素的保护作用使得胰岛素稳定性好,在冻干和复溶过程中减少了胰岛素的降解或变性造成的活性损失。另外还测试了大豆磷脂(spc)和二油酸甘油酯(gdo)分别以32:68、50:50、以及70:30的比例混合后样品的流变性能,采用流变仪考察凝胶的弹性模量(g’)与粘性模量(g”),实验结果见图2,可以看出随着spc/gdo比例增大,所形成的的凝胶弹性模量先增大后减小。另外还考察了不同比例的大豆磷脂(spc)和二油酸甘油酯(gdo)的样品形成凝胶后在不同脂肪酶含量作用下的降解情况。大豆磷脂(spc)和二油酸甘油酯(gdo)分别以35:65、50:50、以及70:30的比例,并且分别添加0%、2%和5%的脂肪酶,测试其重量变化。结果如图3所示。可以看出在脂肪酶的作用下,凝胶先发生吸水溶胀,质量增加,随后降解,质量降低;并且随脂肪酶的含量增多,降解速度加快。综上述结果可以看出本发明制备的亲水性药物的油相液晶凝胶前体制剂中载体在体内可以降解,制剂进入人体后遇水可形成凝胶,凝胶中药物的体外16天的累积释放率均不超过15%,亲水性药物具有长久的缓释,且相对对比例中方法制得的油相制剂和水相制剂可以显著提高亲水性药物的稳定性。尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1