一种基于谱系聚类的古生物谱系演化分析方法与流程
本发明属于生物信息学技术领域,涉及一种构建古生物谱系树的方法。
背景技术:
古生物谱系树的构建是生物信息学重要的组成部分,也是探索生命进化史的主要途径。层次化谱系树细致地展现了物种的演化过程,揭示了物种之间的进化关系和量化差异。早期古生物的谱系分析对生命的起源追溯有着重要的科学意义。谱系分析的目的是找到最简约的谱系树,也就是简约树。
目前,常用于古生物的谱系分析的方法可以分为两大类:1)基于最优原则的方法,包括贝叶斯系统发生推论法(bayesianinference,bi)、最大似然法(maximumlikelihood,ml)和最大简约法(maximumparsimony,mp)等;2)基于距离的方法,包括邻接法(neighborjoining,nj)等。
然而,传统的方法没有考虑到古生物形态学数据矩阵中含有不适用特征状态。由于谱系分析中使用的物种的形态学特征可能在逻辑上依赖的,特征之间存在着包含关系。在古生物学领域中,生物形态的一些复杂特征可以分解为次要特征。这些可分解的复杂特征称为上层特征,由复杂特征分解的次要特征称为下层特征。上层特征和下层特征之间存在包含关系。下层特征仅适用于具有上层特征的物种,没有上层特征的物种在下层特征上被编码为不适用特征状态。特征之间的包含关系导致了不适用特征状态的出现。
目前也已经提出了一些方法来处理演化分析中的不适用数据。这些方法主要有两种,一种是将不适用特征状态视为缺失数据(missingdatareplacement,mdr)。但这种方式将会隐含地对具有该特征的物种加权。不适用特征状态与缺失数据是有本质区别的,即:缺失数据是可能存在的,不适用特征状态是不可能存在。另外一种方式是将不适用特征状态视为一种“自然”特征状态(separatevaluereplacement,svr)。但是这种方法违背该特征上的同源性假设,也会隐含地对不具有该特征的物种加权,而且这种方法仅仅可以在最大简约法中应用。
技术实现要素:
本发明的目的是提供一种准确率高、稳定性好的基于谱系聚类的古生物谱系演化分析方法。
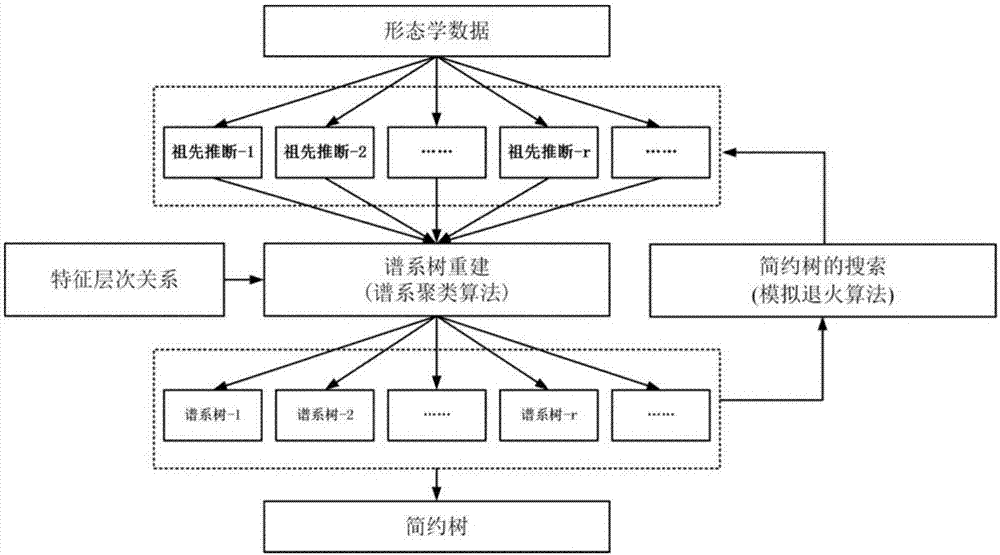
一种基于谱系聚类的古生物谱系演化分析方法,包括以下步骤:
步骤一:建立古生物的特征层次关系模型,并指定特征的演变序列,根据特征演变序列的极向将物种的形态学数据矩阵中的每一维特征状态分为衍征和祖征;
步骤二:根据物种的衍征状态的数目计算物种之间的共近裔指数,选择具有最大近裔指数的物种对生成内节点,计算所述的内节点的特征向量并计算该内节点进化后的谱系树与树长;
步骤三:将内节点的特征向量并加入形态学数据矩阵内,并删除该数据矩阵内中所述的内节点的直接后裔的特征向量,当特征层次模型中只有一个物种,则输出谱系树及其树长,若并非只剩一个物种则转到步骤二;
步骤四:采用模拟退火算法搜索谱系树,得到简约树。
进一步地,步骤一所述的特征演变序列的极向的确定方法包括:
假设物种的形态学数据矩阵为:d{x1,...,xi,...,xj,...,xn},其中xi表示第i个物种;物种xi的第p个形态学特征的状态表示为xip,用于构建谱系树的特征数目记为m;则物种xi的特征向量是xi(xi1,...,xiq,...,xip,...,xim);记第p个特征的上层特征是第q个特征;
通过二进制编码将物种形态学特征的多态转换序列转换为二态转换序列,包括有序的特征多态转换序列和无序的特征多态转换序列,从而确定每一维特征的极向。
进一步地,步骤一所述的将物种的形态学数据矩阵中的每一维特征状态分为衍征和祖征,包括:
令sp是第p个特征的祖征状态,因此,祖先推断s(s0,s1,...,sp,...,sm-1,sm)标记每个特征的祖征状态的一个特征状态集合;以祖先推断为参考,对于物种的形态学数据矩阵中的每一维特征而言,该特征的状态中与祖先推断中标记的该特征的祖征状态相同的特征状态为祖征;否则,剩下的字符状态就是衍征状态。
进一步地,步骤二所述的根据物种衍征状态的数目计算物种之间的共近裔指数的方法为:
共近裔指数d(xi,xj)的计算公式如下所示:
上式中,xip表示第i个物种的第p个特征,xjp表示第j个物种的第p个特征,sp是第p个特征的祖征状态,并且上式中的xip与xjp不属于不可适用特征;
计算每对物种之间的共近裔指数得到共近裔指数矩阵md,共近裔指数矩阵的计算公式如下所示:
进一步地,步骤二所述的选择具有最大近裔指数的物种对生成内节点,包括:
当共近裔指数矩阵md中最大的共近裔指数是唯一的,则选择具有该最大共近裔指数的物种对生成一个内节点;否则,随机选择某个具有最大共近裔指数的物种对一个生成一个内节点。
进一步地,步骤二所述的计算所述的内节点的特征向量并计算该内节点进化后的谱系树与树长,包括:
假设d(xi,xj)是共近裔指数矩阵md中的最大值。因此,选择xi和xj生成一个内节点xk,它是xi和xj的假设祖先,是一个虚拟物种。根据物种xi和xj的特征向量,计算出内节点xk的特征向量。对于xk特征向量中的特征xkp,根据物种xi和xj在第p个特征上的特征状态和第p个特征的祖征状态推断出物种xk在第p个特征上的特征状态,xkq是物种xk第q个特征上的特征状态;
如果在物种xi和xj在第p个特征上的特征状态中不出现不适用特征状态,则根据物种xi和xj在第p个特征上的特征状态xip和xjp,以及第p个特征的祖征状态sp推断出物种xk在第p个特征上的特征状态。如果在物种xi和xj在第p个特征上的特征状态中出现不适用特征状态时,那么xkp的取值由xk的第q个特征的状态决定;
如果xip为衍征状态,而xkp为祖征状态,那么从物种xi到物种xk在第p个特征上发生一次进化事件;从虚拟物种xk进化到物种xi和xj的演化步骤被记为lk(i,j),di表示xi的衍生特征的数目;dk-di、dk-dj分别表示从虚拟物种xk演化到物种xi、xj需要变化的特征状态的数量;在生成一个新的假设祖先之后,从虚拟物种xk到物种xi和xj的演化步骤计算为:
lk(i,j)=(dk-di)+(dk-dj)
l(ts)=∑lk(i,j)
根据祖先推断s进行构建的谱系树表示为ts。树的长度表示为l(ts)。
进一步地,步骤四采用模拟退火算法搜索简约树方法为:
步骤4.1,令初始解状态为s,外循环的迭代次数为l,衰减因子的阈值为β;
步骤4.2,对步骤4.3至步骤4.6进行迭代,使k=1,2…,l;
步骤4.3,产生新解sp',即对于每个特征,我们随机选择一个除了不适用特征状态之外的特征状态(“0”或“1”),作为祖先状态来获得一个新的sp';
步骤4.4,计算增量δf=l(sp')-l(sp),其中l(sp)为评价函数;
步骤4.5,若δf<0则接受sp'作为新的当前解,否则以概率exp(-δf/t)接受sp'作为新的当前解,其中l(sp)和l(sp')之间的变化是衰减因子δf;
步骤4.6,内层循环的终止条件取为δf<β,即连续若干个新解都没有被接受,如果满足终止条件则输出当前解作为最优解,结束程序;
步骤4.7,如果k<l,然后转步骤4.2,反之输出当前谱系树作为简约树。
本发明与现有技术相比具有以下技术特点:
1.本发明为了融入更多演化分析的先验知识以及更加合理地处理演化分析中的不适用数据,提出了一种简约聚类方法进行古生物谱系演化分析。简约聚类包括两个阶段,即谱系树的构建和简约树的搜索。本发明相较于最大简约法等传统构建谱系树的方法能够解决不适用特征状态的造成的数据的不确定性问题,提高了古生物谱系分析的准确率和稳定性。
2.在谱系树的构建阶段,通过融合不对称二元关系和特征空间的层次结构,提出了谱系聚类的方法推断物种之间的演化关系,进而构建谱系树。将性状的极性量化为距离计算以测量特征之间的共享祖先关系,并且根据分层关系推断内部节点的特征向量。因此,在重建谱系树时,不需要关于不可适用数据的同源性假设。
3.在简约树的搜索阶段,在简约原则的基础上采用一种启发式优化算法—模拟退火算法进行简约树的选择。模拟退火算法分解为三部分:解空间,目标函数和初始解。解空间是可能为祖征的每个特征的状态的组合。根据简约原则,需要最小进化步骤来解释状态变化的树是所有可能的谱系树中的最优树。因此,l(ts)是目标函数。我们将每个特征的“0”组成的(0000...0000)设置为初始解,只需sp=(0000...0000)。
附图说明
图1是本发明方法的流程示意图;
图2是谱系聚类的流程图;
图3是寒武纪叶足动物和一些节肢动物分类群的特征层次架构图;
图4是模拟退火算法选择简约树的流程图;
图5是谱系聚类(parsicluster)和最大简约法(mp)对现生物种构建的谱系树的对比图,其中:(a)在陆龟科寄生种数据集(pharyngodonidae)上由谱系聚类(parsicluster)构建的谱系树;(b)在陆龟科寄生种数据集(pharyngodonidae)上由最大简约法(mp)构建的谱系树;(c)在菜花露尾甲属数据集(meligethes)上由谱系聚类(parsicluster)构建的谱系树;(d)在菜花露尾甲属数据集(meligethes)上由最大简约法(mp)构建的谱系树;(e)在木槿数据集(hibiscus)上由谱系聚类(parsicluster)构建的谱系树;(f)在木槿数据集(hibiscus)上由最大简约法(mp)构建的谱系树。在由谱系聚类构建的谱系树和由最大简约法构建的谱系树上有位置不同的物种用粗体标记;
图6是利用邻接法(nj)和谱系聚类(parsicluster)分别构建谱系树,与模型树之间的rf距离的对比图;
图7是传统方法与谱系聚类(parsicluster)分别构建谱系树,与模型树之间的rf距离的对比图。最大简约法(mp-mdr)、最大似然法(ml)和贝叶斯推断(bi)方法中将不适用数据当成缺失数据处理;最大简约法(mp-svr)中将不适用数据当成缺失另一种“自然”状态处理。
具体实施方式
以下结合附图和实施例对本发明进一步的说明。
本发明公开了一种基于谱系聚类的古生物谱系演化分析方法,具体包括以下步骤:
步骤一:根据先验知识建立特征层次关系模型,并指定特征的演变序列,根据特征演变序列的极向将物种的形态学数据矩阵中的每一维特征状态分为衍征和祖征;
步骤1.1,根据先验知识建立古生物的特征层次关系模型,并指定特征的演变序列;
特征层次关系模型是根据古生物学家的先验知识,即古生物学家提供的古生物形态学特征以及特征描述,将特征之间的包含关系进行抽象的一种模型。根据古生物学家提供的外类群(outgroup)等先验知识指定每个特征的演变序列。
在古生物学领域中,生物形态学的一些复杂特征可以分解为次生特征。这些可分解的复杂特征称为上层特征,由复杂特征分解的次要特征称为下层特征。上层特征和下层特征之间存在逻辑依赖关系。
通过对寒武纪叶足动物和一些节肢动物分类群形态学数据集中的特征描述,得到如图3所示的特征层次图。
假设物种的形态学数据矩阵为:d{x1,...,xi,...,xj,...,xn},其中xi表示第i个物种,共有n个物种。物种xi的第p个形态学特征的状态表示为xip,用于构建谱系树的特征数目记为m。因此,物种xi的特征向量是xi(xi1,...,xiq,...,xip,...,xim);假设第p个特征的上层特征是第q个特征,则当某些物种缺少第q个特征所代表的部分时,就会在第p个特征上出现的不适用特征状态。
步骤1.2,通过二进制编码将物种形态学特征的多态转换序列转换为二态转换序列,包括有序的特征多态转换序列和无序的特征多态转换序列,从而确定每一维特征的极向。
其中多态转换序列是古生物形态学特征的各个特征的状态值的转换序列,二态转换序列是当特征的只有两种状态值的转换序列。
步骤1.3,根据特征演变序列的极向将物种的形态学数据矩阵中的特征状态分为两类,即衍征和祖征;
特征的极向是衍征和祖征之间的辩证关系。令sp是第p个特征的祖征状态,因此,祖先推断s(s0,s1,...,sp,...,sm-1,sm)标记每个特征的祖征状态的一个特征状态集合。以祖先推断为参考,对于物种的形态学数据矩阵中的每一维特征而言,该特征的状态中与祖先推断中标记的该特征的祖征状态相同的特征状态为祖征。否则,剩下的字符状态就是衍征状态。
步骤二:根据物种衍征状态的数目计算物种之间的共近裔指数,选择具有最大近裔指数的物种对生成内节点,计算所述的内节点的特征向量并计算该内节点进化后的谱系树与树长;
步骤2.1,根据物种衍征状态的数目计算物种之间的共近裔指数;
共近裔指数d(xi,xj)的计算公式如下所示:
上式中,xip表示第i个物种的第p个特征,xjp表示第j个物种的第p个特征,sp是第p个特征的祖征状态,并且上式中的xip与xjp不属于不可适用特征。
根据系统发育分析的原理,特征的衍征和祖征是不对称的二元关系,物种之间相同的衍征状态的数目被定义为共近裔指数d(xi,xj),该指数用于度量两个物种共享祖先的程度,也就是两个物种亲缘关系的远近。
计算每对物种之间的共近裔指数得到共近裔指数矩阵md,共近裔指数矩阵的计算公式如下所示:
步骤2.2,选择具有最大近裔指数的物种对生成内节点,计算所述的内节点的特征向量并计算该内节点进化后的谱系树与树长;
生成计算内节点特征向量的方法是:当共近裔指数矩阵md中最大的共近裔指数是唯一的,则选择具有该最大共近裔指数的物种对生成一个内节点;否则,随机选择某个具有最大共近裔指数的物种对一个生成一个内节点。
假设d(xi,xj)是共近裔指数矩阵md中的最大值,因此,选择xi和xj生成一个内节点xk,它是xi和xj的假设祖先,是一个虚拟物种。根据物种xi和xj的特征向量,计算出内节点xk的特征向量。对于xk特征向量中的特征xkp,根据物种xi和xj在第p个特征上的特征状态和第p个特征的祖征状态推断出物种xk在第p个特征上的特征状态,xkq是物种xk第q个特征上的特征状态,“-”表示特征的不可适用状态,详细计算过程如下所示:
如果在物种xi和xj在第p个特征上的特征状态中不出现不适用特征状态,则根据物种xi和xj在第p个特征上的特征状态xip和xjp,以及第p个特征的祖征状态sp推断出物种xk在第p个特征上的特征状态;如果在物种xi和xj在第p个特征上的特征状态中出现不适用特征状态时,那么xkp的取值由xk的第q个特征的状态决定。
如果xip为衍征状态,而xkp为祖征状态,那么从物种xi到物种xk在第p个特征上发生一次进化事件,演化步骤是1步。从虚拟物种xk进化到物种xi和xj的演化步骤被记为lk(i,j)。di表示xi的衍生特征的数目。dk-di、dk-dj分别表示从虚拟物种xk演化到物种xi、xj需要变化的特征状态的数量,并代表了演化所需的步骤。在生成一个新的假设祖先之后,从虚拟物种xk到物种xi和xj的演化步骤可以计算为:
lk(i,j)=(dk-di)+(dk-dj)
l(ts)=∑lk(i,j)
根据祖先推断s进行构建的谱系树表示为ts。树的长度表示为l(ts)。
步骤三:将内节点的特征向量加入所述的形态学数据矩阵内,并删除该数据矩阵内中所述的内节点的直接后裔的特征向量;当特征层次模型中只有一个物种,则输出谱系树ts及其树长l(sp),若并非只剩一个物种则转到步骤二;
步骤四:采用模拟退火算法搜索谱系树,得到简约树。
步骤4.1,令初始解状态为s(是算法迭代的起点),外循环的迭代次数为l,衰减因子的阈值为β;
模拟退火算法分解为三部分:解空间,目标函数和初始解。解空间是最可能成为祖征的每个特征的状态,它由sp的所有可能值组成。根据简约原则,需要最小演化步骤来解释状态变化的树是所有可能的谱系树中的最优树。这里l(sp)是评价函数,其表达式为l(sp)=∑lk(i,j),当l(sp)的取值越小则说明算法越优。我们将每个特征的“0”组成的(0000...0000)设置为初始解,只需sp=(0000...0000);
步骤4.2,对步骤4.3至步骤4.6进行迭代,使k=1,2…,l;
步骤4.3,产生新解sp',即对于每个特征,我们随机选择一个除了不适用特征状态之外的特征状态(“0”或“1”),作为祖先状态来获得一个新的sp';
步骤4.4,计算增量δf=l(sp')-l(sp),其中l(sp)为评价函数;
步骤4.5,若δf<0则接受sp'作为新的当前解,否则以概率exp(-δf/t)接受sp'作为新的当前解,其中l(sp)和l(sp')之间的变化是衰减因子δf;
步骤4.6,内层循环的终止条件取为δf<β,即连续若干个新解都没有被接受,如果满足终止条件则输出当前解作为最优解,结束程序;
步骤4.7,如果k<l,然后转步骤4.2,反之输出当前谱系树作为简约树。
为了验证本方法的有效性,本发明选取已发表论文中的生物形态学数据进行了实验验证:
实验选取陆龟科寄生种等6个生物形态学数据集作为实验数据集。首先,为了验证本发明提出的方法在不含不适用数据集上的效果,本文提出的方法与最大简约法进行了比较,实验结果如图5所示。实验结果表明,谱系树的绝大部分分支是相同的,只有少数物种位置不一样。其次,由于本发明提出的方法属于距离法,因此将本发明提出的方法与邻接法进行了比较,实验结果如图6所示。实验结果表明,本发明提出的方法在谱系树构建方面明显优于邻接法。最后,为了验证本发明提出的方法在古生物数据集上的效果,即数据集中含有大量不适用数据,将本发明提出的方法与目前可行的所有用于处理不适用数据的方法进行对比,包括贝叶斯推断,最大简约法和最大似然法等,实验结果如图7所示。实验结果表明,本发明提出的方法在古生物形态学数据集上具有一定的优势。
- 还没有人留言评论。精彩留言会获得点赞!