叶酸受体结合配体-药物偶联物的制作方法
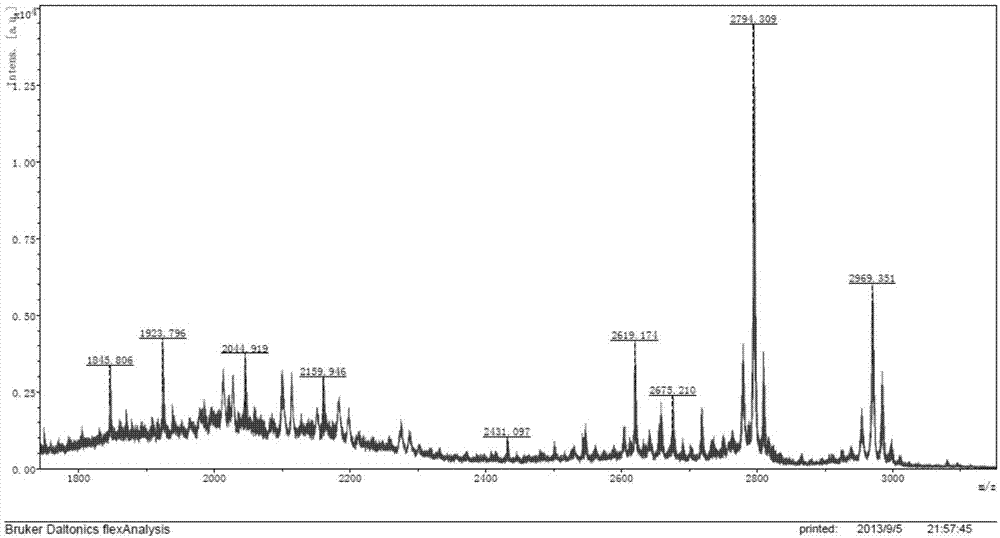
本发明涉及用于靶向递药的药物化合物及其制备方法。具体涉及叶酸受体结合配体-药物偶联物,更具体的,本发明涉及两个或两个以上叶酸受体结合配体(f)通过接头(l)与药物或其类似物或衍生物(d)偶联,形成(f)nld偶联物,本发明也涉及,所述偶联物用于治疗由病原性细胞群体引起的疾病状况,以及涉及这样的偶联物的制备方法和其药物组合物。发明背景尽管癌症的治疗方法和抗癌药物的研究已经取得了很大进展,但癌症仍是严重威胁人类健康的主要疾病之一,在美国,癌症是继心脏病之后第二大导致死亡的原因。现在治疗癌症的代表性方法包括外科疗法、放射线疗法、化学疗法、免疫疗法及以上治疗方法联合应用对抗癌症。最经常地,利用高强效药,例如丝裂霉素、紫杉醇和喜树碱,通过化疗法治疗癌症。其中化疗药物的主要缺点在于抑制肿瘤细胞生长的同时,也严重抑制了正常细胞的增殖,这些不可避免的副作用使得新的特异性抗癌药物的开发显得尤为重要。目前已经进行许多努力通过将抗癌药物结合到配位体如激素、抗体、或维生素来发展肿瘤选择性药物。例如,将低分子量维生素化合物(如叶酸)用作肿瘤靶向剂。叶酸(fa)也称维生素b9,是所有活细胞维持一碳途径正常代谢和核苷酸生物合成所需的必须营养物。叶酸受体(fr)是一种跨膜单链糖蛋白,含有3种亚型:α-fr、β-fr、γ-fr。叶酸(fa)对叶酸受体(fr)的细胞表面定向的糖蛋白表现出极高的亲和力(kd~100pm),该糖蛋白是从细胞外环境中捕捉其配体(叶酸)的糖基磷脂酰肌醇连接的蛋白。叶酸受体(fr)是与叶酸(fa)结合的肿瘤相关膜蛋白且能够在细胞内通过胞吞机制转运与叶酸结合的分子。结合之后,包围fr-配体复合物的质膜将立即内陷以形成内部囊泡,称作内体。囊泡腔的ph通过共定位内体膜中的质子泵的作用稍微变低,且这种酸化作用可能介导着fr蛋白中的构象改变以释放其所结合的配体从而允许进入胞质。研究表明在90%的卵巢癌、晚期乳腺癌、宫颈癌、子宫内膜癌、结肠癌、肺癌、脉络膜癌、室管膜细胞瘤中出现高表达的α-fr;在恶性增殖的白细胞(白血病)表面及类风湿关节炎等自身免疫性疾病患者体内激活的巨噬细胞表面有高表达的β-fr,而叶酸受体在正常组织中几乎不表达。因此叶酸和叶酸受体在靶向治疗技术中具有很好的研究前景,尤其是在肿瘤和自身免疫性疾病的治疗方面,fr作为抗肿瘤药物的靶点受到极大的关注,成为新型抗肿瘤药物研究的热点之一(hilgenbrink,a.andp.low(2005).folatereceptormediateddrugtargeting:fromtherapeuticstodiagnostics.journalofpharmaceuticalsciences94(10):2135-2146.)。国际上已经在尝试利用叶酸-放射性核显像剂偶联物,如叶酸与125i、67ga、111in的偶联物能非侵入性的探测人体高表达叶酸受体的肿瘤组织。同时,目前国际上已广泛就叶酸-蛋白毒素、叶酸-小分子化疗药物、叶酸-脂质体(脂质体内包化疗药物或基因药物)及叶酸-免疫治疗剂等展开了深入研究(参考文献:hilgenbrink,a.andp.low(2005).folatereceptormediateddrugtargeting:fromtherapeuticstodiagnostics.journalofpharmaceuticalsciences94(10):2135-2146。wo2012065085、wo2012065079、us2012022245a1)。技术实现要素:本发明目的是提供一种对致病细胞具有特异性并对正常细胞毒性小的叶酸受体结合配体-药物偶联物,其制备方法以及其在制备治疗肿瘤药物中的应用。在本发明的一个说明性实施方式中,描述了具有以下化学式的化合物:(f)nld的叶酸受体结合配体-药物偶联物,其中n为2、3或4;f是叶酸受体结合配体,选自叶酸或蝶酸;(f)n中每个f独立的选自叶酸和蝶酸;d是药物或其类似物或衍生物;l是包含以下结构的接头:l1-l2其中,l1是多价接头,l2包含至少一个可释放接头,l1与l2共价连接;应当理解的是,本发明中所述(f)n是指每个f分别与多价接头l1共价连接,d与l2共价连接。在另一实施方式中,描述了具有以下结构的叶酸受体结合配体-药物偶联物:其中f1、f2独立的选自叶酸和蝶酸;d是药物或其类似物或衍生物;l是包含以下结构的接头:l1-l2其中,l1是多价接头,l2包含至少一个可释放接头,l1与l2共价连接;所述的f1、f2分别与多价接头l1共价连接,d与l2共价连接。所述的多价接头(l1)可含有相互共价连接的多个接头,包括一种或多种间隔接头、可释放接头、杂原子接头及其以任何次序排列的组合。在一可变化方式中,可释放接头和任选的间隔接头互相共价结合为形成接头l1;在另一个可变化方式中,配体f通过一个或多个间隔接头共价连接到多价接头l1;在另一个可变化方式中2个或2个以上配体f直接相互附着或通过多个间隔接头和/或可释放接头共价附着;在另一可变化方式中,两个或更多个可释放接头相互共价连接,并且其中一个或多个可释放接头通过杂原子接头和/或间隔接头相互隔开。应当理解的是,一种或多种可释放接头和任选的间隔接头的排列的可变性很大。所述的杂原子接头中的杂原子为氮、氧、硫、磷、硅等。进一步的,所述杂原子接头(氧除外)可为不同氧化态,如n(oh)、s(o)、s(o)2、p(o)、p(o)2、p(o)3等。在另一可变化方式中,所述杂原子接头为羟胺、肼、腙、磺酸酯、亚瞵酸酯、瞵酸酯等。在一个实施方案中,多价接头l1包含至少一个由氨基酸形成的肽间隔接头,所述的氨基酸独立的选自天然氨基酸和非天然α-氨基酸。在另一个实施方案中,所述多价接头l1包括含有1~40个氨基酸形成的肽间隔接头,所述氨基酸优选天然氨基酸,进一步的,所述多价接头l1包括含有1~20个氨基酸形成的肽间隔接头;更进一步的,所述l1优选包括含有10~15个氨基酸形成的肽间隔接头。更进一步的,l1包括至少2个选自下组的氨基酸:天冬氨酸、精氨酸、半胱氨酸、赖氨酸、天冬酰胺、精氨酸、苏氨酸、谷氨酸、丝氨酸、瓜氨酸、缬氨酸和谷氨酰胺。更进一步的,所述l1优选包括一个或多个由选自天冬氨酸、精氨酸、半胱氨酸、瓜氨酸、缬氨酸和赖氨酸及其组合的氨基酸形成的二肽、三肽、四肽、五肽、六肽、七肽、八肽、十肽、十一肽及十二肽的间隔接头。在一个示例性的实施方式中,由氨基酸形成的肽间隔接头与杂原子接头组合在一起形成如下式的多价接头l1:其中,*表示开放价。所述的多价接头l1任选的包括选自如下的间隔接头:聚醚、糖、硫代羰基、亚烷基、1-亚烷基琥珀酰亚胺-3-基、1-(羰基烷基)琥珀酰亚胺-3-基、羰基烷基羰基、1-(羰基四氢-2h-吡喃基)琥珀酰亚胺-3-基和1-(羰基四氢呋喃基)琥珀酰亚胺-3-基,其中所述每个间隔接头任选被一个或多个取代基取代;其中所述的取代基独立的选自是烷基、烷氧基、烷氧基烷基、羟基、羟烷基、氨基、氨烷基、烷基氨烷基、二烷基氨烷基、巯基烷基、烷基硫代烷基、芳基、取代的芳基、芳烷基、取代的芳烷基、杂芳基、取代的杂芳基、羧基、羧基烷基、羧酸烷基酯、链烷酸烷基酯、胍基烷基、或由氨基酸及其衍生物和肽取代的羰基或酰氨基或酰氨基烷基。本发明中,l2包含至少一个可释放接头,所述“可释放接头”是指包括至少一个在生理条件下可断裂的键(例如ph不稳定键、酸不稳定键、氧化不稳定或酶不稳定键)的接头。不言而喻,这种导致键断裂的生理条件包括发生在生理ph下的,或作为区室化入细胞器,例如其ph比胞质ph低的核内体的结果而发生的标准化学水解反应。不言而喻,如本发明所述,可断裂键可连接可释放接头内的两个相邻原子,和/或在所述可释放接头的任一端或两端连接其它接头或配体f和/或药物d。在这种可裂解键连接可释放接头内的两个相邻原子的情况下,该键断裂后,所述可释放接头断裂成两个或更多个片段。或者,在这种可裂解键在可释放接头和另一部分例如杂原子接头、间隔接头、另一可释放接头、药物或其类似物或衍生物或叶酸受体结合配体之间的情况下,该键断裂后,可释放接头从其它部分中分离出来。可裂解键的不稳定性可进行调整,例如通过在可裂解鍵位置或其附近的取代改变,例如包括毗邻可裂解二硫键的α分支化,同系化形成可水解的部分缩酮或缩醛的烷氧基等。所述的可释放接头选自:1-烷氧基亚烷基、1-烷氧基亚烷基羰基、1-烷氧基亚环烷基羰基、双羰基芳基、双羰基羧基芳基、双羰基双羧基芳基、卤代亚烷基羰基、氧基羰基氧基、氧基羰基氧基烷基、亚氨基烷基、羰基亚烷基亚氨基、亚氨基亚环烷基、羰基亚环烷基亚氨基、亚烷基硫基、亚烷基芳基硫基和羰基烷基硫基,其中每个所述可释放接头任选被一个或多个取代基取代;其中所述的取代基独立的选自是烷基、烷氧基、烷氧基烷基、羟基、羟烷基、氨基、氨烷基、烷基氨烷基、二烷基氨烷基、巯基烷基、烷基硫代烷基、芳基、取代的芳基、芳烷基、取代的芳烷基、杂芳基、取代的杂芳基、羧基、羧基烷基、羧酸烷基酯、链烷酸烷基酯、胍基烷基、或由氨基酸及其衍生物和肽取代的羰基或酰氨基或酰氨基烷基。优选的,本发明所述化合物中,所述可释放接头包括二硫化物、碳酸酯、酰肼、酰胺、腙、氨基酸酯、脲或其组合。进一步优选的,本发明所述可释放接头包括一个或多个具有如下结构的式子:其中,n为0、1、2、3或4;r为h,烷基、任选取代的酰基或氮保护基;x为o、ch2或nh;y为o或s,z为nh、o或s;r1为烷基、羧基取代烷基或酰基取代烷基;*表示开放价。更进一步的,由所述可释放接头与杂原子接头或间隔接头形成具有如下结构的接头l2:其中,m为0~4的整数,w选自nh或o;*表示开放价。授权公告号为cn100381177c的专利详细描述了可释放接头和间隔接头,所述文献的公开内容通过引用结合到本发明中。所述多价接头l1与包含可释放接头的l2任选通过一个或多个间隔接头、杂原子接头、可释放接头连接形成接头l。示例性的由l1与l2直接共价连接或通过一个或多个间隔接头、杂原子接头、可释放接头连接形成的接头l包含如下的结构:其中,w选自nh或o;*表示开放价。本发明所提供的叶酸受体结合配体-药物偶联物中,包含叶酸受体结合配体(f),接头(l)和药物(d),可根据本发明提到的任何方式形成接头l或可使用本领域认已知的间隔接头、可释放接头与杂原子接头间通过共价连接来形成接头l,叶酸受体结合配体f或药物d与杂原子接头的连接可通过存在于已经转化成杂原子接头的药物d或叶酸受体结合配体f上的反应性官能团制成。其中,所述叶酸受体结合配体f上的反应性官能团为羧基或氨基,多价接头可接连到所述叶酸受体结合配体f上的羧基或氨基形成酯或酰胺;其中所述药物包括双键氮原子时,可释放接头接连到所述药物氮上形成腙;所述药物可包括硫原子时,所述可释放接头选自亚烷基硫基或羰基烷基硫基,并且可释放接头连接到所述药物的硫上形成二硫键;所述药物可包括氧原子,所述可释放接头为被取代基取代或未被取代的亚烷氧基羰基或卤代亚烷基羰基,所述可释放接头连接到药物的氧上形成碳酸酯或酯;所述药物d可包含氮原子,可释放接头为卤代亚烷基羰基或取得的卤代亚烷基羰基,所述可释放接头连接到药物d的氮上形成酰胺。间隔接头和可释放接头,及杂原子接头可以以不同的方式组合。示例性的说明,所述接头通过杂原子接头相互连接,例如亚烷基-氨基-亚烷基羰基,其中,x、y分别为1~5的整数。示例性的说明性,在另一实施方案中,描述了下式的化合物:其中,w为nh或o;m为0或1;f1、f2独立的选自下式:本发明中所述药物d可为任何能够调节或以另外方式改变细胞功能的分子,包括药物活性化合物。所述的药物活性化合物可为本领域已知药物或其衍生化形式,所述药物为细胞毒性、提高肿瘤通透性、抑制肿瘤细胞增殖、促进细胞凋亡、降低包细胞中抗凋亡活性的药物。适于本发明的药物包括但不限于激素,抗生素,抗微生物药,抗病毒药,抗癌药。本身是细胞毒性的或可用来增加肿瘤通透性的化疗药,也在本发明方法中使用。所述的细胞毒性的药物例如包括cbi(环丙基苯并[e]吲哚酮)类似物或其衍生物,开环-环丙基苯并[e]吲哚酮类似物、o-ac-开环-环丙基苯并[e]吲哚酮类似物、多拉司他汀(dolastatin)类(如多拉司他汀-10),欧瑞抑制素(auristatin)类(如一甲基欧瑞抑制素e(mmae)、一甲基欧瑞抑制素f(mmaf)),管溶解素类(tubulysins)、考布他汀,美登素、美登素类似物、dm1、埃博霉素、紫杉醇及紫杉醇衍生物(如多西他赛)、长春碱及其类似物(如长春新碱、脱乙酰长春碱一酰肼(davlbh))、喜树碱以及喜树碱衍生物、秋水仙碱、异秋水仙碱、硫代秋水仙碱、秋水仙胺、道诺霉素、根霉素、环磷酰胺、氨甲喋呤、博来霉素、替西罗莫司(temsirolimus)、丝裂霉素、微管抑制剂、吡咯并苯并二氮杂卓(pbd)二聚体类、环丙基苯并[e]吲哚酮、开环-环丙基苯并[e]吲哚酮、卡里奇霉素(calicheamicin)。其他可适用于本发明的药物包括大环内酯类抗肿瘤药、化疗药如烷化剂,氮芥、亚硝基脲、白消安、卡铂、苯丁酸氮芥、顺铂和其它铂化合物,抗代谢药如阿糖胞苷,嘌呤类似物,嘧啶类似物及青霉素、头孢菌素、万古霉素、红霉素、克林霉素、利福平、氯霉素、氨基糖苷类抗生素和阿昔洛韦、三氟尿苷、更昔洛韦、齐多夫定、金刚烷胺、利巴韦林吉西他滨和任何其它本领域认可的抗微生物化合物。进一步的,本发明所述药物优选替西罗莫司(temsirolimus)、开环-环丙基苯并[e]吲哚酮类似物、吡咯并苯并二氮杂卓(pbd)二聚体类、卡里奇霉素(calicheamicin)、7-乙基-10-羟基喜树碱(sn-38)、长春碱类、多拉司他汀(dolastatin)、欧瑞抑制素(auristatin)类、膜海鞘素b(didemninb)、管溶解素b(tubulysinb)、美登素类、紫杉醇类、柔红霉素、多柔比星、表柔比星、埃斯波霉素或放线菌素。更进一步的,本发明所述药物优选开环-环丙基苯并[e]吲哚酮类化合物、pbd二聚体类、卡里奇霉素、sn-38、davlbh、tubulysinb、didemninb、mmae、mmaf、mmaf衍生物、dm1、紫杉醇类、长春新碱、柔红霉素、多柔比星或表柔比星。公开号为wo2009/064913a1和us2010113476a1的专利对cbi类化合物及开环-环丙基苯并[e]吲哚酮类化合物进行了详细介绍,其公开的部分,通过引用结合到本发明中,示例性的开环-环丙基苯并[e]吲哚酮类化合物如下式所示:其中r1为h、ome、oh、onhboc、onhac、onh(ac)boc、onphth或r2为nh2或ome。所述的吡咯并苯并二氮杂卓(pbd)二聚体类化合物中pbd是指具有如下单元的结构:pbd二聚体类化合物的区别在于芳环a和吡咯环c两者的取代基的数目、类型和位置以及吡咯环c的饱和度不一样,示例性的pbd二聚体类化合物如:其中,r1为羟基、氨基、c1~c6的烷氧基、c1~c6的烷基、c1~c6的氨基取代的烷基、c1~c3的醛基或羰基、或c1~c6环烷基或杂环基;r2是2-、3-或4-取代,为-n=nh-或-nh-;n为0~3的整数。公开号为cn201180029867的专利对此类化合物进行了详细描述,并公开了其制备方法,在其中公开的内容通过引用结合到本发明中。所述sn-38是具有下式的结构:所述davlbh是具有下式的结构:tubulysinb和mmae具体结构如下所示:mmaf及其衍生物是指具有如下结构的化合物:其中,ry为c1~c6的烷基或卤代烷基或任意取代的含1~4个碳原子的羰基;当ry为氢时,即为mmaf。所述didemninb是具有下式的结构:其中,rx为对甲氧基苯基。所述dm1是具有下式的结构:所述叶酸受体结合配体偶联物中,药物d与可释放接头接头l2的连接,可通过存在药物d或其衍生化形式的反应性官能团制成,例如didemninb的羟基转化成相应的碳酸酯,另一个说明性实施例是,tubulysinb的末端羧基先衍生化为酰肼,然后酰肼上的氮原子作为杂原子接头再与l上的可释放接头连接,叶酸转化成相应的酰胺等,如下式所说明:下述叶酸受体结合配体-药物偶联物为说明性的本发明提供的药物偶联物,认为它们在本发明所述范围内,这些叶酸-药物偶联物可根据本领域认可的方案或本发明所述方法制备。在另一个实施方案中,本发明描述了如下结构的化合物:其中,rx为对甲氧基苯基;x1为cl或br;r为h、ome、oh、onhboc、onhac、onh(ac)boc、onphth或ry为氢、c1~c6的烷基或卤代烷基或任意取代的含1~4个碳原子的羰基;r1为c1~c6的烷氧基或烷基或氨基取代的烷基、或c1~c3的醛基或羰基、羟基、氨基或c1~c6环烷基或杂环基;r2是苯的2-、3-或4-位取代,为n=nh或nh;n为0~3的整数。其中onphth表示以下结构:进一步的,本发明提供了如下结构的化合物:其中,l4选自以下的结构:f1,f2独立的选自下式:*表示连接位置。进一步的,本发明提供了如下式的化合物:另一方面,本发明提供了具有如下通式的化合物:f3ld及f4ld,其中f、l、d之间的连接方式类似于化合物f2ld中f、l、d之间的连接方式。示例性的f3ld及f4ld化合物如下所示:其中,d选自前文所述的药物或其衍生物或类似物;f1、f2、f3、f4独立的选自:l2是接头,包含至少一个可释放接头,任选的包含一个或多个杂原子接头、间隔接头。示例性的,l2具有如下结构:其中,m为0~4的整数,w选自nh或o;*表示开放价。进一步的,以下具体化合物用以说明f3ld及f4ld:本发明所述的叶酸受体结合配体-药物偶联物可通过本领域认可的合成方法制备。所述合成方法的选择取决于杂原子接头的选择,药物的结构特点,以及所述间隔接头和所述可释放接头上官能团。一般而言,相关键形成反应描述于richardc.larock.comprehensiveorganictransformations,aguidetofunctionalgrouppreparations.vchpublishers,inc.newyork(1989),和theodorae.greene&peterg.m.wuts.protectivegroupsionorganicsynthesis.第二版,johnwiley&sons,inc.newyork(1991),所述文献的公开部分通过引用结合到本发明中。其中所述杂原子接头为硫原子,存在于所述可释放接头的官能团为亚烷基硫醇衍生物,所述二硫化物基团可通过相应的杂芳基二硫基衍生物如吡啶-2-基二硫基烷基衍生物等与亚烷基硫醇衍生物反应形成。其反应溶剂可选择四氢呋喃(thf)、n,n-二甲基甲酰胺(dmf)、二氯甲烷、二甲基亚砜(dmso)等,反应温度在0℃~80℃之间变动。常规碳酸酯、硫代碳酸酯和氨基甲酸酯的形成,通常可通过使羟基取代的化合物、硫基取代的化合物或胺取代的化合物分别与活化烷氧基羰基衍生物反应,形成碳酸酯、硫代碳酸酯和氨基甲酸酯。反应可在常规溶剂如thf、dmf、dmso、二氯甲烷、乙酸乙酯等中进行,反应温度在0~80℃范围变动,可使用任何碱性催化剂如无机碱、胺碱、聚合物结合碱等促进该反应。常用酰胺和酯的形成,例如,其中所述杂原子接头为氮原子,存在于间隔接头或可释放接头上的末端官能团为羰基,所述的酰胺基可通过相应的羧基或其衍生物酯化反应或酰化反应得到,成酰胺反应溶剂包括二氯甲烷、thf、dmf、dmso等,说明性的所述酰胺可在-15~80℃变动范围内进行反应制备。偶联剂包括dcc、edc、hbtu、tbtu、hobt/dcc、hobt/edc等,或者母体酸可被转化成活化的羰基衍生物,例如酰氯、n-羟基琥珀酰亚胺基酯等,成酰胺反应也可在碱性条件下如三乙胺、n,n'-二异丙基乙胺等中进行。同样的,所述杂原子接头为氧原子,存在间隔接头或可释放接头上的末端基团为羰基,所需要的酯基可通过相应的羧酸或其衍生物与醇偶联反应制备。所述偶联剂包括dcc、edc、cdi、bop、eedq、dead、三苯基膦等,溶剂包括二氯甲烷、thf、dmf、dmso、乙腈、乙酸乙酯等,碱包括三乙胺、二异丙基乙胺等。或者母体酸可被转化成活化的羰基衍生物,例如酰氯、n-羟基琥珀酰亚胺基酯等。所述药物包括氮原子,可释放接头或间隔接头可以接连到所述药物d的氮原子上形成腙,所需要的腙基可通过相应的醛或酮和肼或酰肼衍生物反应形成。可用溶剂包括二氯甲烷、thf、dmf、dmso、氯仿、乙酸乙酯等,反应温度在0~80℃范围变动,可用任何酸性催化剂如矿物酸、冰醋酸、三氟乙酸等作为催化剂。酰腙可通过合适的羧酸或其衍生物酰化肼,然后使酰肼与相应的醛或酮反应形成酰腙。或者,所述腙官能团可通过使肼与相应的醛或酮反应形成。所得腙随后再用合适的羧酸或其衍生物酰化。常规的琥珀酰亚胺的形成:所述杂原子接头为氮原子、氧原子或硫原子,存在与间隔接头或可释放接头上的官能团为琥珀酰亚胺衍生物,所得的碳-杂原子可通过相应胺、醇或硫醇和顺丁烯酰亚胺衍生物的迈克尔加成形成。形成迈克尔加成的溶剂包括thf、乙酸乙酯(etoac)、ch2cl2、dmf、dmso、h2o等。这个种迈克尔加成化合物的形成可用加入等摩尔量的碱如三乙胺或通过调节水溶液的ph至6.0~7.4来完成。显而易见的,当所述杂原子接头为氧或氮原子时,可调节反应条件来易化迈克尔加成,如通过采用较高反应温度、加入催化剂、使用极性较强的溶剂如dmf、dmso等,以及用硅烷基化试剂活化顺丁烯二酰亚胺。对称缩醛或缩酮基可由相应的醇和醛或酮通过缩醛和缩酮反应而形成。一般遵循r.r.schmidt等,chem.rev.,2000,100,4423-42所综述的步骤,所述文献的公开内容通过引用结合到本发明中。常规的叶酸-肽的制备:通过聚合物-载体的顺序方法,用fmoc-策略,在对酸敏感的2cl-trtresin树脂上制备含叶酸的肽基片段pte-γglu-(aa)n-cys-oh,如下方案所示:(a)20%哌啶/dmf;(b)fmoc-aa-oh,hobt,dic,dmf;(c)fmoc-glu-otbu,hobt,dic,dmf;(d)n10-tfa-pteroicacid;pybop,dipea,dmf/dmso;(e)2%nh2nh2/dmf;(f)tfa/h2o/苯酚/茴香硫醚/edt(82.5:5:5:5:2.5);在本发明所述方法的说明性实施方式中,r1为fmoc,r2三苯基甲基,dic为二异丙基碳二亚胺,dipea为n,n'-二异丙基乙胺,为确保有效偶联,用pybop作活化剂。在标准条件下,在每个偶联步骤后,将fmoc保护基除去。按照scheme1中所述,使用适当保护的氨基酸构件例如fmoc-glu-otbu、n10-tfa-pteroicacid等,并以步骤(b)中fmoc-aa-oh为代表。因此,aa是指任何适当保护的氨基酸原料。应当理解的是,本发明所用的术语“氨基酸”是指任何具有一个或多个碳分隔的胺或羧酸官能团,并包括天然存在的α和β氨基酸,以及这些氨基酸的衍生物和类似物。具体地,具有侧链的被保护氨基酸例如被保护的丝氨酸、苏氨酸、半胱氨酸、天冬氨酸等也可用于本发明所述叶酸-肽的合成。此外,γ、δ或更长的同源侧链或替代的分支结构的氨基酸类似物,例如正亮氨酸、异缬氨酸、β-甲基苏氨酸、β-甲基半胱氨酸、β,β-二甲基半胱氨酸等也可作为原料包括在本发明所述叶酸-肽的合成中。涉及fmoc-aa-oh的偶联序列(步骤(a)和(b))进行n次,以制备固体支持肽(ii),其中n为整数并可为0~100。最后的偶联步骤之后,除去剩余的fmoc基(步骤(a)),所述肽随后被偶联至谷氨酸衍生物(步骤(c)),脱保护,并偶联至tfa-保护的蝶酸(步骤(d))。所述tfa保护基通过用碱处理(步骤(e))除去,再经步骤(f)得到含肽基片段(iii)。然后,所述肽通过用三氟乙酸(tfa)、h2o、苯酚、茴香硫醚和edt处理,从聚合物载体上切下(步骤(f))。这些反应条件导致同时去除t-bu、t-boc、pbf和trt保护基,所述保护基可形成适当保护的氨基酸侧链的部分。含三或四个叶酸肽的片段可按照类似的方法制备,其中的多价接头可通过含有3个或3个以上反应性官能团(如氨基、羟基、羧基)的氨基酸构建,所述的氨基酸例如可以是赖氨酸、谷氨酸、丝氨酸、天冬酰胺、天冬氨酸、酪氨酸、谷氨酸精氨酸等。示例性的含三个叶酸肽的片段iii-2可按照如下方案制备:先将两个赖氨酸连接,得到含有四个反应性官能团的化合物c,再将化合物c加入反应柱,茚三酮监测,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid,(其中,氨基酸和hobt/dic的用量分别为15eq和16.5eq/16.5eq)缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得到iii-2。示例性的含四个叶酸肽的片段iii-3可按照如下scheme3所示方法制备:类似的,先将四个赖氨酸进行缩合,得到化合物f,在将化合物f加入反应柱,茚三酮监测,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid,(其中,氨基酸和hobt/dic的用量分别为15eq和16.5eq/16.5eq)缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得到目标化合物iii-3。其中c和f中的任何一个赖氨酸也可用其他结构类似的氨基酸或取代的氨基酸替代,用以制备类似含有四个或五个反应性官能团的片段,并且不同氨基酸的缩合顺序、连接方式及氨基酸的立体构型并不用于限制本发明。n10-tfa-pteroicacid是指具有如下结构的化合物:其制备方法具体参见题目为“efficientsynthesesofpyrofolicacidandpteroylazide,reagentsfortheproductionofcarboxyl-differentiatedderivativesoffolicacid”(j.am.chem.soc.,vol.119,no.42,1997,10004-10013)的文献,该文献公开了由叶酸制备n10-tfa-pteroicacid的具体方法,其公开的部分通过引用结合到本发明中。在另一个方案中,本发明提供了一种药物组合物,其包含在此描述的叶酸受体结合配体-药物偶联物和药学上可接受的载体、稀释剂或赋形剂、或其组合。在另一个方案中,本发明提供了一种所述药物组合物在治疗和/或预防由致病细胞群体引起的疾病的用途。所述致病细胞群体是指癌细胞、感染性物质如细菌和病毒、被细菌或病毒感染的细胞、能够引起病症的活化的巨噬细胞,以及任何独特的、优先表达或过度表达叶酸受体的其他类型致病细胞。所述致病细胞群体可为致瘤(包括良性肿瘤和恶性肿瘤)的癌细胞群体,或可为不致瘤的。所述的癌细胞群体包括但不限于,口腔癌、甲状腺癌、内分泌腺癌、皮肤癌、胃癌、食道癌、喉癌、胰腺癌、结肠癌、膀胱癌、骨癌、卵巢癌、子宫癌、乳腺癌、睾丸癌、前列腺癌、直肠癌、肾癌、肝癌和肺癌。根据本发明,所述叶酸受体结合配体-药物偶联物可用于治疗以宿主中存在致病细胞群体为特征的疾病,其中致病细胞群体的成员具有叶酸或蝶酸结合部位,其中所述结合部位被所述致病细胞独特表达、过量表达或优先表达。所述致病细胞的选择性消除,由所述叶酸受体结合配体-药物偶联物的维生素部分连接到叶酸受体。高亲和力叶酸受体表面表达维生素受体在肿瘤细胞上过量表达。卵巢、乳腺、结肠、肺、鼻、咽和脑上皮癌都被报道表达高水平叶酸受体。事实上,已知超过90%的人类卵巢肿瘤以高水平表达这种受体。因此,本发明递药缀合物可用于治疗各种肿瘤细胞类型,以及其它致病细胞类型如感染因子,所述细胞类型优先表达叶酸受体,并因此具有表面可及的维生素或维生素类似物或衍生物结合部位。本发明中使用的术语“叶酸盐或叶酸、蝶酸盐或蝶酸”是独立的指代用于形成偶联物的叶酸,或蝶酸部分。缩写:“boc”表示叔丁氧羰基;“cdi”表示n,n-羰基二咪唑;“ddq”表示2,3-二氯-5,6-二氰基-1,4-苯醌;“dcc”表示n,n'-二环己基碳二亚胺;“dead”表示偶氮二甲酸二乙酯;“dipea”和“diea”均表示n,n'-二异丙基乙胺;“dic”表示n,n'-二异丙基碳二亚胺;“edci”表示1-(3-二甲基氨基丙基)-3-乙基碳二亚胺;“edc”表示(1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐);“eedq”表示2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉;“fmoc”表示芴甲氧羰基;“hbtu”表示(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基四氟硼酸脲鎓;“hatu”表示n-[[(二甲基氨基)-1h-1,2,3-三唑并[4,5-b]吡啶-1-基]亚甲基]-n-甲基六氟磷酸甲铵n-氧化物;“hoat”表示7-氮杂-1-羟基苯并三唑;“hobt”表示1-羟基苯并三唑;“hosu”表示n-羟基琥珀酰亚胺;“ivdde”表示1-(4,4-二甲基-2,6-二氧代环亚己基)-3-甲基丁基;“nmp”表示n-甲基吡咯烷酮;“otbu”表示叔丁氧基;“paba”表示对氨基苯甲酸;“tbtu”表示2-(1h-苯并噻唑基)-1,1,3,3-四甲基脲四氟硼酸;“tbme”表示叔丁基甲醚。本发明提供的叶酸受体结合配体-药物偶联物中叶酸受体结合配体为2个或更多个,优选2~4个,并且所述每个配体分别与多价接头共价连接,药物d与可释放接头共价连接,与现有技术中只含有一个受体结合的配体相比,本发明提供的偶联物与叶酸受体细胞亲和力极大的提高,并且本发明所述的多价接头l包含可释放接头,当偶联物的叶酸和/或蝶酸部分结合到致病细胞上,偶联物与致病细胞表面紧密结合,随后内吞到细胞内,在细胞内可释放接头迅速裂解,将药物d释放,发挥药物的药效作用,出乎意料的是,本发明提供的偶联物在叶酸受体过表达的肿瘤细胞中表现出显著的抗肿瘤活性,而基于动物体重变化得出的结果发现,本发明所提供的偶联物在显著提高抗肿瘤活性的同时,在所给药物剂量下,动物均能很好的耐受。并且,与现有的叶酸受体结合配体-药物偶联物(如ec145)相比,同等摩尔剂量下,本发明提供的偶联物与叶酸受体的结合几率更大;更出乎意料的是,即使是在过量叶酸存在条件下,同等摩尔剂量时,与现有的叶酸受体结合配体-药物偶联物(如ec145)相比,本发明提供的偶联物表现出较稳定的抗肿瘤活性,这表明,即使在存在一定量的叶酸的竞争条件下,本发明提供的偶联物也能发挥很好的抗肿瘤活性,本发明的这些优点可以通过后面的具体实施例进一步得到证明。与现有技术中的叶酸受体结合配体-药物偶联物相比,本发明提供的偶联物分子量显著的增大,有利于偶联物通过被动靶向机制选择性的聚集在肿瘤细胞表面,同时本发明所提供的偶联物含有2个或多个叶酸受体结合配体,进一步增强了偶联物与叶酸受体阳性表达的肿瘤细胞的亲和力以及偶联物在肿瘤细胞中的滞留时间,进一步的,与药物共价连接的可释放接头如酰肼键以及接头l中的其他可释放接头如二硫键在细胞酸性条件下,几分钟内酰肼键就断开,同时二硫键被还原,释放药物,迅速发挥药物的抗肿瘤活性。此外,发明人还发现,本发明所提供的偶联物与血浆白蛋白结合率低,这也预示着本发明所提供的偶联物在体内清除速率快,毒性小。以下结合附图和具体实施例对本发明的有益效果进一步说明,以下实施例仅为说明性,并不对本发明进行限制。附图说明图1显示由实施例4制备的化合物15的质谱图,其中2969.35为化合物15的分子离子峰。图2显示1μmbp111b(本发明的化合物15)及对照品bp111a(化合物ec145)在存在和不存在过量叶酸条件下对kb细胞的抑制作用,其中“-”表示不加入,“+”表示加入。图3显示bp111b(本发明的化合物15)及对照品bp111a(化合物ec145)对叶酸受体阴性的a549细胞生长的抑制作用,横坐标表示加入bp111a和bp111b的浓度。图4显示分别给予0.5μmol/kg、1μmol/kg、2μmol/kgbp111b(化合物15)和对照品bp111a对小鼠kb肿瘤生长的抑制作用,vehicle表示空白对照,即:不给予药物治疗,仅给予等量溶媒,纵坐标tv(mm3)表示肿瘤体积(tumorvolume),横坐标:天(day)。图5显示分别给予0.5μmol/kg、1μmol/kg、2μmol/kgbp111b(化合物15)和对照品bp111a对kb肿瘤小鼠体重影响,vehicle表示空白对照,即:不给予药物治疗,仅给予等量溶媒,纵坐标bw(g)表示体重(bodyweight),横坐标:天(day)。具体实施例以下通过具体实施例对本发明的技术方案及优点进一步说明,认为它们在本发明所述范围内,并不作为对本发明的限制。所述方法为说明性方法,可用于制备本发明所述的药物缀合物。实施例1化合物iii的制备根据本发明所述的常规步骤,按照scheme1的方案,向50ml反应瓶中,加入1000mg2cl-trtresin(1.5eq),再加入234mgfmoc-cys(trt)-oh(1.0eq),8ml二氯甲烷(dcm),640μldiea(2.0eq),完全溶解,反应50min后加入8mldcm、1mlmeoh、1mldiea反应20min后,转移至自制固相合成反应柱中,dmf洗涤后加入10ml20%的哌啶dmf溶液,反应20min,dmf清洗后茚三酮监测阳性,取709mgfmoc-lys(fmoc)-oh(3.0eqiv,178mghobt(3.3eqiv),6mldmf溶解后加入230μldic(3.3eq)混合后加入反应柱,反应1h后茚三酮监测,dmf洗涤后加入20%的哌啶dmf溶液。重复以上步骤,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid(其中,氨基酸和hobt/dic的用量分别为6eq和6.6eq/6.6eq),缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得粗品iii,经hplc制备色谱分离纯化后,得到纯品iii,ms[m]+:2097.74。实施例2化合物33的制备向100ml三口烧瓶中加入10.0mldcm,1.0mlmeoc(o)scl(1.0eq)冰浴降温至0℃,滴加0.76ml巯基乙醇(1.0eq),保温0℃反应30min,加入1.22g2-巯基吡啶(1.0eq),16mldcm滴加进反应瓶,保温0℃反应1h后升至室温1h后tlc原料反应完毕,后处理:浓缩至16ml左右,抽滤,滤液用dcm洗涤后得淡黄色微臭味固体2.0g。向100ml三口烧瓶中加入3.0mldcm、0.46g双光气(0.55eq),冰浴降温至0℃,将1.0g化合物33-2(1.1eq)溶于13mldcm中,再向其中加入0.45g三乙胺(1.0eq),溶解后滴加到反应瓶中,滴加时控温0℃以下,升至室温1h后tlc原料反应完毕(即得33)收率96%,ms[m]+:248.97。该方法的其他细节见美国专利us2007276018和i.r.vlahov,etal.bioorg.med.chem.lett.16(2006):5093–5096所述,这两篇文献通过引用整体结合到本发明中。实施例3化合物15-3的制备向100ml三口烧瓶中加入1.5g长春碱、5ml无水甲醇和5ml无水肼,升温至约60℃,反应24h后tlc检测反应进程,反应结束后,倒入50ml水中,dcm萃取3次,水洗3次,饱和食盐水洗3次,无水硫酸钠干燥浓缩后得白色固体1.1g,即化合物15-2。向100ml三口烧瓶中加入化合物15-20.98g(1.0eq),化合物330.67g(1.5eq)、10gdcm,搅拌溶解后向反应瓶中滴加0.44ml三乙胺(2.5eq),室温2h后反应完毕。后处理:加入50mldcm,水洗3次,饱和食盐水洗3次,无水硫酸钠干燥后浓缩得粗品1.2g,柱层析后得15-3的纯品750mg。ms[m]+:959.43。实施例4化合物15的制备向100ml三口瓶中加入7.5ml水,氮气鼓泡30min后加入200mg化合物iii(1.2eq),用0.1n的nahco3水溶液调节ph约为6.9;将78mg化合物15-3溶于15mlmeoh中,并将其加入到反应瓶中,反应约10min后反应完毕,hplc制备后冻干得化合物15的纯品39mg。hplc:98.1%,采用基质辅助激光解析电离飞行时间质谱(maldi-tof-ms)测得化合物15的质谱图见附图1,ms[m+h]+:2969.35。实施例5化合物16的制备类似化合物15的制备方法制备化合物16。ms[m+h]+:3043.18。实施例6化合物17的制备类似化合物15的制备方法制备化合物17。ms[m+h]+:2819.98。实施例7化合物18的制备(rx为对甲氧基苯基)将化合物33与dmap(4-二甲基氨基吡啶)加入到didemninb的二氯甲烷溶液中在0℃反应2h,分离纯化后得到化合物18-1;将化合物iii溶于水中,氮气条件下,用碳酸氢钠调节溶液ph为7,将化合物18-1溶于乙腈中,并在氮气条件下,加入到化合物iii的水溶液中,利用反相分析hplc检测反应进程,反应结束后,分离纯化,得到目标产物18,ms[m+h]+:3312.36。实施例8化合物19的制备在0℃条件下处于5%甲醇/二氯甲烷中的化合物19-1(7.7mg,10μmol)的混合物中加入化合物19-2的三氟乙酸盐(6.4mg,20μmol)。将反应混合物升温至环境温度并搅拌5h,然后减压浓缩,硅胶柱层析(洗脱剂:3%甲醇/二氯甲烷),得到3.3mg目标化合物19-3,ms[m+h]+:974.47。将化合物19-3与化合物iii的巯基进行迈克尔加成反应,制备获得化合物19,ms[m+h]+:3058.16。实施例9化合物20的制备化合物20的制备可按照上述路线由化合物20-1经过8步反应制备获得;其中,由化合物20-1经过6步反应制备化合物20-9的具体操作方法在ep0624377a2中有详细说明,该文献的公开部分通过引用结合到本发明中。化合物20-10的制备:mmae(100.5mg,0.14mmol,1eq),化合物20-9(110.6mg,0.15mmol,1.1eq)及hobt(19mg,0.14mmol,1.0eq)用dmf(2ml)溶解。2min后,加入吡啶(0.5ml),用反相hplc监测反应进程。反应约24h后,反应结束,将反应液浓缩,残留物用反相制备hplc纯化(variandynamax色谱柱21.4mm×25cm,5μ,100孔),使用乙腈和et3n-co2(ph7)从10%到100%进行梯度洗脱,流速为20ml/min,洗脱时间为40min。分段收集,浓缩,得到类白色固体,为化合物20-10,ms[m]+:1315.78。将化合物20-10与化合物iii的巯基进行迈克尔加成反应,得到化合物20。ms[m+h]+:3414.52。实施例10化合物21的制备参照实施例9的方法制备化合物21。ms[m+h]+:3428.50。实施例11化合物22的制备本发明所使用的马来酰亚胺修饰的缬氨酸-瓜氨酸二肽(化合物22-1)的合成方法参考文献:genem.d.,etal.cathepsinb-labiledipeptidelinkersforlysosomalreleaseofdoxorubicinfrominternalizingimmunoconjμgates:modelstudiesofenzymaticdrμgreleaseandantigen-specificinvitroanticanceractivity.bioconjugatechem.13(4)855-869(2002)。化合物22-1与cbi的反应按照常规的氨基与羧基反应制备酰胺的方法进行操作,得到化合物22-2,类似化合物19的制备方法,将化合物22-2与化合物iii的巯基反应,得到化合物22。ms[m+h]+:3065.16。实施例12化合物23的制备在吡啶条件下按照本领域常规方法用(boc)2o将sn-3810位的羟基进行保护得到化合物23-2,将化合物23-2(0.358g,0.073mmol),dmap(0.266,0.218mmol)及三光气(0.0095g,0.032mmol)加入到反应瓶中,然后加入1.5ml二氯甲烷引发反应,tlc检测反应进程,反应结束后用无水甲醇淬灭,得到化合物23-3,不经处理直接用于下一步反应。将化合物20-8(0.768g,0.883mmol)加入到上述23-3的反应液中,反应约5min反应结束,用快速色谱柱纯化(甲醇/二氯甲烷梯度洗脱),得到20位被boc保护的化合物23-4,然后再用tfa水解脱去保护基,用乙醚重结晶得到目标化合物23-4的tfa盐。按照scheme6的方案,化合物23-4与化合物iii的半胱氨酸的-sh反应制备得到目标化合物23。ms[m+h]+:3089.15。实施例13化合物24的制备类似化合物15的制备方法制备化合物24。ms[m+h]+:2946.23。实施例14化合物25的制备类似化合物15的制备方法制备化合物25。ms[m+h]+:2932.20。实施例15化合物26的制备类似化合物15的制备方法制备。ms[m+h]+:2918.22。实施例16化合物27的制备根据本发明所述的常规步骤,按照scheme1的方案,向50ml反应瓶中,加入1000mg2cl-trtresin(1.5eq),再加入234mgfmoc-cys(trt)-oh(1.0eq),8ml二氯甲烷(dcm),640μldiea(2.0eq),完全溶解,反应50min后加入8mldcm、1mlmeoh、1mldiea反应20min后,转移至自制固相合成反应柱中,dmf洗涤后加入10ml20%的哌啶dmf溶液,反应20min,dmf清洗后茚三酮监测阳性,取709mgfmoc-lys(fmoc)-oh(3.0eqiv,178mghobt(3.3eqiv),6mldmf溶解后加入230μldic(3.3eq)混合后加入反应柱,反应1h后茚三酮监测,dmf洗涤后加入20%的哌啶dmf溶液。重复以上步骤,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,n10-tfa-pteroicacid,(其中,氨基酸和hobt/dic的用量分别为6eq和6.6eq/6.6eq),缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得iii-1,ms[m]+:1839.65。类似实施例4中化合物15的制备方法,制备化合物27,不同之处是用iii-1替代实施例4中iii。ms[m+h]+:2711.05。实施例17化合物iii-1’全保护肽asp(otbu)-asp(otbu)-arg(pbf)-asp(otbu)-pteroicacid的合成:采用2cl-trtresin,spps依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,n10-tfa-pteroicacid,缩合完毕后加入2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干。加入1%的tfa/dcm溶液全保护切割5min,重复数次,tlc直至没有产物为止,滤液用吡啶中和后水洗,饱和食盐水洗涤后浓缩,柱层析的到全保护肽asp(otbu)-asp(otbu)-arg(pbf)-asp(otbu)-pteroicacid纯品,ms[m]+:981.05。iii-1’的合成:向50ml反应瓶中,加入1000mg2cl-trtresin(2.0eq),再加入117mgfmoc-cys(trt)-oh(1.0eq),8mldcm,320μldiea(2.0eq),完全溶解,反应50min后加入8mldcm,1mlmeoh,1mldiea,反应20min后转移至自制固相合成反应柱中,dmf洗涤后加入10ml20%的哌啶dmf溶液,反应20min,dmf清洗后茚三酮监测阳性,取fmoc-lys(ivdde)-oh(3.0eq),178mghobt(3.3eq),6mldmf溶解后加入230μldic(3.3eq)混合后加入反应柱,反应1h后茚三酮监测,dmf洗涤后加入20%的哌啶dmf溶液。重复以上步骤,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid,(其中,氨基酸和hobt/dic用量分别为3eq和3.3eq/3.3eq),缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,茚三酮监测阳性,加入化合物{asp(otbu)-asp(otbu)-arg(pbf)-asp(otbu)-pteroicacid}(2.0eq),hoat(2.2eq),dic(2.2eq),dmf做溶剂。反应过夜后茚三酮监测,反应完毕后dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得iii-1’。ms[m]+:1968.69。实施例18化合物28的制备类似实施例4中化合物15的制备方法,制备化合物28,ms[m+h]+:2840.10。实施例19化合物iii-2的制备按照scheme2所示的方法制备iii-2,具体步骤如下:化合物c的制备:250ml反应瓶中加入10ga,1.1eq的hosu,100ml的thf,溶解后冰浴降温至0℃,加入1.1eq的dcc升至室温搅拌过夜。tlc原料反应完毕。后处理:抽滤,滤饼用thf洗涤,有机相浓缩后得12g化合物b。100ml反应瓶中加入3.0g的d,溶于30ml水,加入1.1eq的nahco3,降温至10度;将1.1eq的b溶于30mldme,滴加进反应瓶,补加10mlthf室温过夜。tlc反应完毕,浓缩后加入ea,稀盐酸洗涤,然后水洗,食盐水洗,干燥,浓缩,柱层析得到化合物c。iii-2的制备:向50ml反应瓶中,加入1000mg2cl-trtresin(2.0eqiv),再加入117mgfmoc-cys(trt)-oh(1.0eq),8mldcm,320μldiea(2.0eq),完全溶解,反应50min后加入8mldcm,1mlmeoh,1mldiea,反应20min后转移至自制固相合成反应柱中,dmf洗涤后加入10ml20%的哌啶dmf溶液,反应20min,dmf清洗后茚三酮监测阳性,取化合物c(3.0eq),178mghobt(3.3eq),6mldmf溶解后加入230μldic(3.3eq)混合后加入反应柱,反应1h后茚三酮监测,dmf洗涤后加入20%的哌啶dmf溶液。重复以上步骤,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid,(其中,氨基酸和hobt/dic用量分别为15eq和16.5eq/16.5eq),缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得iii-2,ms[m+h]+:3151.15。实施例20化合物iii-3的制备按照scheme3所示的方法制备iii-2,具体步骤如下:化合物f的制备:250ml反应瓶中加入10ga,1.1eq的hosu,100ml的thf,溶解后冰浴降温至0度,加入1.1eq的dcc升至室温搅拌过夜。tlc原料反应完毕。后处理:抽滤,滤饼用thf洗涤,有机相浓缩后得12g化合物b。向100ml反应瓶中,加入3.0g的d,溶于30ml水,加入1.1eq的nahco3,降温至10℃;将1.1eq的b溶于30mldme,滴加进反应瓶,补加10mlthf室温过夜。tlc反应完毕,浓缩后加入ea(乙酸乙酯),稀盐酸洗涤,然后水洗,食盐水洗,干燥,浓缩,柱层析得到化合物c。向10ml反应瓶中加入5gc,1.1eq的hosu,50ml的thf,溶解后冰浴降温至0℃,加入1.1eq的dcc升至室温搅拌过夜。tlc原料反应完毕。后处理:抽滤,滤饼用thf洗涤,有机相浓缩后得6g化合物e。向100ml反应瓶中加入3.0g化合物e,溶于30ml水,加入1.1eq的nahco3,降温至10℃;1.1eq的b溶于30mldme,滴加进反应瓶,室温过夜。tlc反应完毕,浓缩后加入ea,,稀盐酸洗涤,然后水洗,食盐水洗,干燥,浓缩,柱层析得到化合物f。iii-3的制备:向50ml反应瓶中,加入1000mg2cl-trtresin(2.0eq),再加入117mgfmoc-cys(trt)-oh(1.0eq),8mldcm,320μldiea(2.0eq),完全溶解,反应50min后加入8mldcm,1mlmeoh,1mldiea反应20min后转移至自制固相合成反应柱中,dmf洗涤,然后加入10ml20%的哌啶dmf溶液,反应20min,dmf清洗后茚三酮监测阳性,取化合物f(3.0eq),178mghobt(3.3eq),6mldmf溶解后,加入230μldic(3.3eq)混合后加入反应柱,反应1h后茚三酮监测,dmf洗涤后加入20%的哌啶dmf溶液。重复以上步骤,依次缩合fmoc-asp(otbu)-oh,fmoc-asp(otbu)-oh,fmoc-arg(pbf)-oh,fmoc-asp(otbu)-oh,fmoc-glu-otbu,n10-tfa-pteroicacid,(氨基酸和hobt/dic分别为15eq和16.5eq/16.5eq)缩合完毕后加入10ml2%水合肼/dmf溶液反应5分钟,重复3次,dmf洗涤,dcm洗涤,meoh洗涤,抽干,合成完毕,加入裂解试剂,tbme沉淀多肽,得到目标化合物iii-3,ms[m+h]+:4203.55。实施例21化合物29的制备类似实施例4中化合物15的制备方法,制备化合物29,不同之处是用iii-2替代实施例4中iii。ms[m+h]+:4021.54。实施例22化合物30:ms[m+2h]2+:4481.91。化合物31:ms[m+2h]2+:5074.95。化合物32:ms[m+2h]2+:5520.33。试验实施例1相对亲和力试验将fr阳性kb细胞密集种植在20孔细胞培养板上,并允许所述细胞粘附于塑料持续18h。将消耗过的培养基置于指定的孔中,在提高和不提高受试品或叶酸浓度的情况下用100nm3h-叶酸补充不含叶酸的rpmi(ffrpmi)。将细胞在37℃培养60min,然后用ph7.4的pbs漂洗3次。向每孔加入500μl的含1%十二烷基磺酸钠(sds)的ph7.4的pbs溶液。然后,收集细胞裂解液并将其加入至含有5ml荧光混合物的单独管中,然后计数其放射性。阴性对照管只含有溶于ffrpmi中的叶酸(不含竞争者)。阳性对照管含有终浓度为1mm的叶酸,并且这些样品中测得的cpm(代表标记的非特异性结合)从所有样品中扣除。显而易见地,相对亲和力被定义为置换50%的结合于kb细胞上fr的3h-叶酸所需化合物的反摩尔比(inversemolarratio),叶酸对fr的相对亲和力被设为1。化合物15在10%血清/fdrpmi中的相对亲和力试验结果表明,与叶酸相比,化合物15表现出157%的叶酸受体相对亲和力。类似的方法,测定化合物16~32在10%血清/fdrpmi中的相对亲和力,试验结果显示,与叶酸相比,化合物16~32均表现出超过100%的叶酸受体相对亲和力,其中化合物20、21、31分别表现出162%、127%和187%的叶酸受体相对亲和力。试验实施例2细胞活性试验细胞活性试验1:本发明化合物15~32用体外细胞毒性试验评价,该试验预测药物抑制叶酸受体阳性的kb细胞生长的能力,每孔中接种100μl含kb细胞5*103个的pbs溶液,培养24h后,吸取介质,分成空白对照组、对照组a、试验组b,对照组a和试验组b分别分为10组,编号为a-1、a-2、a-3、a-4、a-5、a-6、a-7、a-8、a-9、a-10和b-1、b-2、b-3、b-4、b-5、b-6、b-7、b-8、b-9、b-10,对照组和试验组分别依次加入叶酸(fa)0、0.1、0.3、1、3、10、30、100、300微摩尔(μm),继续培养2h,然后向对照组a分别加入对照化合物bp111a(化合物ec145,ec145的制备方法及结构见中国专利cn100381177c)1μm,向试验组b加入本发明的化合物(用bp111b表示)1μm,空白对照组既不加药物也不加叶酸;继续培养70h,细胞存活率用mtt法评价,在酶联免疫检测仪od570nm处测量各孔的吸光值。mtt法又称mtt比色法,其检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt还原为水不溶性的蓝紫色结晶甲瓒(formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(dmso)能溶解细胞中的甲瓒,用酶联免疫检测仪在5700nm波长处测定其光吸收值(即od570值),可间接反映活细胞数量。在一定细胞数范围内,mtt结晶形成的量与细胞数成正比。举例说明,以化合物15作为试验组b的药物(用bp111b表示),按照上述方法操作,试验结果如图2所示,其中bp111b表示本发明的化合物15,bp111a表示化合物ec145(ec145的制备方法见中国专利cn100381177c)。从图2可看出,在相同条件下,bp111b(本发明的化合物15)对kb细胞的抑制作用优于化合物bp111a(化合物ec145),并且,随着fa的增加,bp111b对kb肿瘤细胞的抑制作用降低,这表明,所观察到的细胞杀伤作用是通过结合于叶酸受体介导的。并且,与bp111a相比,bp111b即使是在过量叶酸存在的条件下,对kb肿瘤细胞也具有较好的抑制活性,也如图2所示。细胞活性试验2:该试验预测药物抑制叶酸受体阴性的a549细胞生长的能力,每孔中接种100μl含a549细胞5*103个的pbs溶液,培养24h后,吸取介质,分成空白对照组、对照组a、试验组b,对照组a和试验组b分别分为10组,编号为a-1、a-2、a-3、a-4、a-5、a-6、a-7、a-8、a-9、a-10和b-1、b-2、b-3、b-4、b-5、b-6、b-7、b-8、b-9、b-10,对照组a和试验组b分别加入对照化合物bp111a(ec145,ec145的制备方法见中国专利cn100381177c)和试验化合物bp111b10000、3333.33、111.111、370.370、123.4567、41.152、13.717、4.572、13.717、4.5724、1.5241纳摩尔(nm),空白对照组既不加bp111a也不加bp111b;继续培养70h,细胞存活率用mtt法评价,在酶联免疫检测仪od570nm处测量各孔的吸光值。以化合物15作为试验组药物bp111b为例,试验结果如图3所示,bp111b(本发明化合物15)对fr-阴性的肿瘤细胞a549抑制活性不明显,这些结果表明化合物15是通过叶酸选择性的或叶酸特异性的机理发挥作用。对本发明所述化合物16~32在这类型的试验中都得到类似的结果,在多数情况下,本发明所提供的化合物16~32对kb细胞具有显著的抑制活性,并且呈现剂量依赖的细胞毒性,其ic50(ic50即半抑制率,意思是产生50%肿瘤生长抑制作用的时候药物的浓度)均在低纳摩尔范围内,如下表1所示。表1化合物16~32对kb细胞抑制活性数据(ic50:nm)化合物ic50(nm)化合物ic50(nm)161.4251.7172.2261.21810.2271.9191.8281.7202.1291.2212.6301.3225.9310.9233.8320.8241.1试验实施例3对小鼠肿瘤生长的抑制试验本发明所述化合物15(用bp111b表示)静脉内(i.v.)给予荷瘤动物时,在带有皮下kb肿瘤的小鼠的balb/c小鼠上评价了其抗肿瘤活性。在右腋皮下组织用1*106kb细胞肿瘤接种后约11天(t0时平均肿瘤体积=60mm3),对小鼠分成9组,每组小鼠为6只,其中a-1、a-2、a-3组为药物对照组a;b-1、b-2、b-3为试验组b;剩余三组为空白对照组。对照组a分别静脉内注射给予0.5μmol/kg、1μmol/kg、2μmol/kg对照药物bp111a;试验组b分别静脉内注射给予0.5μmol/kg、1μmol/kg、2μmol/kg化合物bp111b(本发明化合物15);空白组给予相当剂量体积的pbs,每周静脉注射3次,持续2周,每个治疗组每周2次,用测径规测量肿瘤生长。用方程v=axb2/2计算肿瘤体积,其中,a为肿瘤长度,b为宽度,单位为毫米。同时每周2次或3次用天平称量动物体重。如图4和5所示,用本发明所述化合物bp111b0.5μmol/kg治疗时,即可有效延迟kb肿瘤的生长,药物剂量增加到1μmol/kg时具有非常明显的抗肿瘤活性。并且,如图4所示,用本发明所述化合物bp111b对肿瘤的抑制作用呈剂量依赖性,与对照药物bp111a相比,同样给药剂量下,本发明化合物bp111b表现出更好的抗肿瘤活性。而且本发明提供的化合物bp111b没有明显毒性(基于动物体重),与对照药物bp111a相比,同样给药1μmol/kg时,对照药物bp111a也具有抗肿瘤活性,但是在给药18天时,bp111b对肿瘤的抑制作用明显优于bp111a,如图4所示;而基于动物体重变化发现,bp111b和bp111a一样,在所给药剂量下,动物均能很好的耐受,如图5所示。本发明化合物16~32进行类似的试验,试验结果发现:使用本发明的化合物16~32对fr阳性的小鼠kb肿瘤生长具有显著的抑制作用,并且没有明显伴有体重变化的毒性。说明性的,化合物25或游离的药物mmaf或相当剂量体积的pbs(对照)以500nmol/kg的剂量给在右腋皮下组织用1*106kb细胞肿瘤接种后约11天(t0时平均肿瘤体积=60mm3)小鼠(6只/组)静脉内注射,每周静脉注射3次,持续2周后,观察对小鼠肿瘤生长的抑制结果和小鼠体重变化。每个治疗组每隔3天,用测径规测量肿瘤生长。用方程v=axb2/2计算肿瘤体积,其中,a为肿瘤长度,b为宽度,单位为毫米。同时每隔3天用天平称量动物体重。实验结果发现,用本发明所述化合物25治疗能有效延迟kb肿瘤的生长,而且没有明显毒性(基于动物体重)。游离药物mmaf也具有抗肿瘤活性,但是具有明显的毒性,且动物耐受性差。类似方法,测定化合物25在fr-阴性细胞中的抗肿瘤活性,在右腋皮下组织用1*106a549细胞肿瘤接种后(t0时平均肿瘤体积=50~80mm3),对小鼠(6只/组)静脉内注射给予500nmol/kg的化合物25或相当剂量体积的pbs(对照),每周静脉注射3次,持续2周,每个治疗组每隔3天,用测径规测量肿瘤生长。用方程v=axb2/2计算肿瘤体积,其中,a为肿瘤长度,b为宽度,单位为毫米。试验结果表明,化合物25对fr-阴性细胞几乎没有活性,这些结果表明化合物25是通过叶酸选择性的或叶酸特异性的机理发挥作用。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 5‑HT2B拮抗剂的制造方法与工艺
- 1‑(噻唑‑2‑基)哌啶‑4‑醇的合成方法与制造工艺
- 一种大麻酚类化合物的合成方法与制造工艺
- 一类对GPR84具有激动作用的化合物及其制备方法和用途与制造工艺
- eGFP标记GPR120阳性细胞的小鼠模型构建方法
- 靶向生长抑素受体的Al<sup>18</sup>F?NOTA?PEG<sub>6</sub>?TATE及其制备方法和应用
- 吡咯烷衍生物的口服制剂的制作方法
- 胰高血糖素样肽-1受体激动剂制备治疗肺动脉高压药物的应用
- 一种armc5基因检测试剂盒及突变位点的制作方法
- 作为mglu5受体的负性别构调节剂的6-氟-2-[4-(吡啶-2-基)丁-3-炔-1-基]咪唑并[1,2a ...的制作方法