一种RNA测序数据的基因表达量计算方法与流程
本发明属于基因表达数据分析技术领域,具体涉及一种rna测序数据基因表达量计算方法。
背景技术:
对基因表达数据进行分析是分子生物学的核心手段,近年来,随着高通量测序技术的发展,rna-seq技术成为了测定基因表达量的主要手段,海量的原始rna-seq数据得到了积累,这些公开的数据对于研究者而言具有十分重要的意义。然而,将原始rna-seq序列读段(reads)转化为定量化的基因表达数据这一过程涉及了很多的专业软件,例如序列的质量控制,序列的比对和组装等。这些软件的使用需要一定的生物信息学背景和编程知识,这给使用rna-seq数据带来了一定的门槛。因此,很多研究学者都视图降低这一门槛,目前的工具有两种,一种是对公开的rna-seq数据进行收集和处理,提供整合的基因表达定量数据库,该方法的缺点是需要花费大量的人力和物力,因此主要集中在生物医疗方面的rna-seq数据,此外,该方法具有一定的时滞性,通常没有囊括最新发表的数据;另一种是提供软件服务,允许用户直接对符合其研究兴趣的rna-seq数据进行下载和定量化,然而,这种方法仍然涉及了很多专业的参数和技术性细节需要用户花费时间学习和设置;另外以上两种方法都只允许用户利用公开发表的rna-seq数据,不能帮助他们对自己的测序数据进行定量化,再者,目前主要存在两种基于rna-seq定量基因表达量的方案:依赖参考序列对比(alignment-based)或者不依赖参考序列比对(alignment-free),两种方案各有优缺点;目前不论是整合的数据库或者是软件服务都只采用了一种方案,这一方面可能会降低rna-seq技术的准确性,另一方面也给横向比较基因的表达量带来了障碍。
技术实现要素:
针对现有技术中的上述不足,本发明提供的rna测序数据的基因表达量计算方法解决了上述背景技术中的问题。
为了达到上述发明目的,本发明采用的技术方案为:一种rna测序数据的基因表达量计算方法,包括以下步骤:
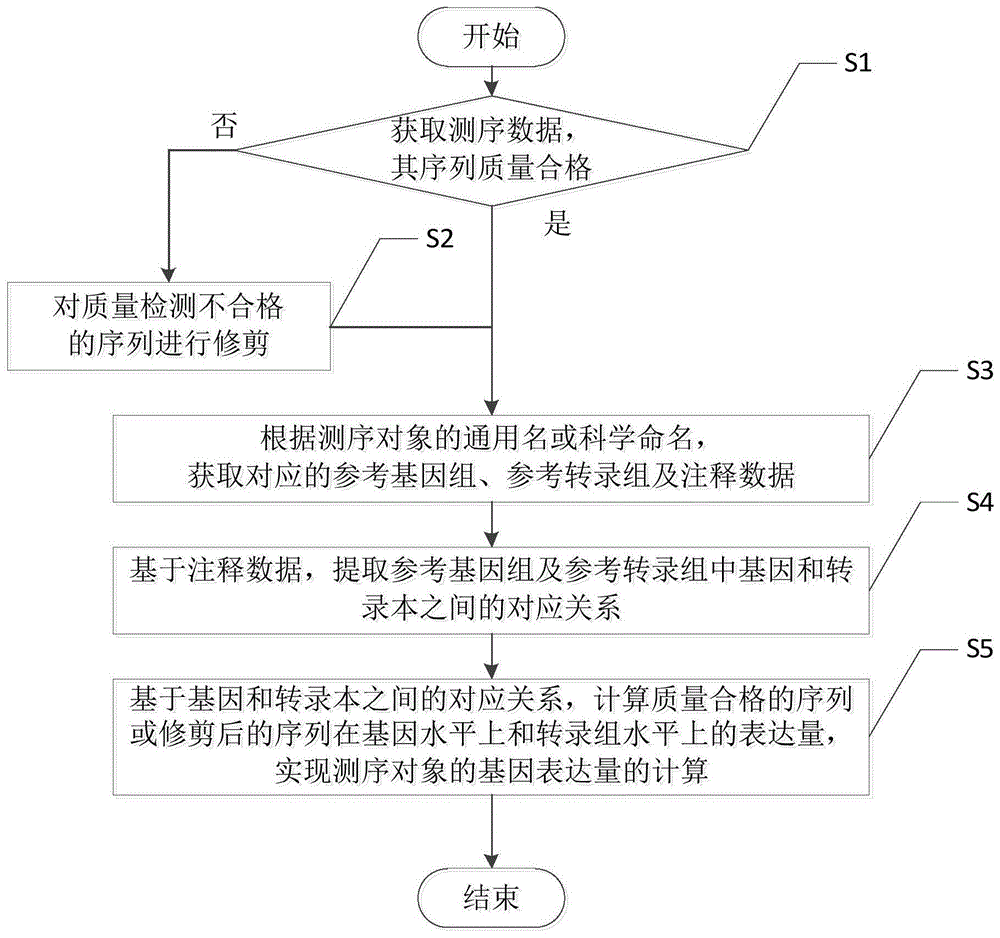
s1、获取rna测序数据,并对其序列进行质量检测,判断序列质量是否合格;
若是,则进入步骤s3;
若否,则进入步骤s2;
s2、对质量检测不合格的序列进行修剪,进入步骤s3;
s3、根据测序对象的通用名或科学命名,获取对应的参考基因组、参考转录组及注释数据,进入步骤s4;
s4、基于注释数据,提取参考基因组及参考转录组中基因和转录本之间的对应关系;
s5、基于基因和转录本之间的对应关系,计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现测序对象的基因表达量的计算。
进一步地,所述步骤s1中,rna测序数据包括用户构建的测序数据和公开的测序数据;
当获取的rna测序数据为公开的测序数据时,将其转换成fastq格式文本,fastq格式文本保存有测序序列及其测序质量得分信息,用于对公开的测序数据的序列进行质量检测。
进一步地,所述步骤s1中,对序列进行质量检测包括测序质量检测和测序接头检测,当测序质量检测不合格或测序接头检测不合格时,序列的质量检测不合格;
进行测序质量检测时,基于测序质量得分信息,当序列中存在测序质量小于设定阈值的碱基或该序列子集的平均测序质量小于设置阈值时,测序质量检测不合格;
进行测序接头检测时,当序列中有超过设定阈值的读段含有测序接头时,测序接头检测不合格。
进一步地,进行测序接头检测时,检测的测序接头包括illumina3端接头、illumina5端接头、illuminauniversal接头、nexteratransposasesequence接头和solid接头。
进一步地,所述步骤s2中,对质量检测不合格的序列进行修剪包括去除序列中低于设定阈值的碱基或去除超过设定阈值的测序接头;
进一步地,所述步骤s4中,在基因和转录本之间的对应关系中,一个基因对应一个或多个转录本。
进一步地,所述步骤s5具体为:
a1、基于基因和转录本之间的对应关系,利用hisat2将质量合格的序列或修剪后的序列对比到参考基因组中对其进行定位,将其保存并转化为bam格式;
a2、利用stringtie将bam格式的序列组装成转录本,并以gtf格式保存;
a3、基于gtf格式文本及基因和转录本之间的对应关系,利用alignment-based方法计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现基因表达量的计算。
进一步地,所述步骤a3中,基于gtf格式文本,质量合格的序列或修剪后的序列在转录组水平上的表达量的计算公式为:
count转录本=coverage*length/read_len
式中,count转录本是转录本的数量,即基因表达量,coverage为转录本的覆盖度,length为转录本的长度,read_len为序列读段的平均长度;
基于基因和转录本之间的对应关系,质量合格的序列或修剪后的序列在基因水平上的表达量的计算公式为:
count基因=∑(count转录本)
式中,count基因为某个基因的表达量,count转录本为基因转录的某个转录本的数量。
进一步地,所述步骤s5具体为:
b1、基于基因和转录本之间的对应关系,利用salmon将质量合格的序列或修剪后的序列伪对比到参考转录组,将其保存为quant.sf格式纯文本;
b2、基于quant.sf格式纯文本,利用alignment-free方法计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现基因表达量的计算。
进一步地,所述步骤b2中,将quant.sf格式纯文本转换为csv格式,得到质量合格的序列或修剪后的序列在转录组水平上的表达量;
所述质量合格的序列或修剪后的序列在基因水平上的表达量的计算公式为:
count基因=∑(count转录本)
式中,count基因为某个基因的表达量,count转录本为基因转录的某个转录本的数量。
本发明的有益效果为:
本发明提供的rna测序数据的基因表达量的计算方法,仅需要两个非专业性的参数就可以自动地生成基于alignment-based和alignment-free的基因表达量,可以在提高基因表达量的定量精度的同时方便用户与其他的研究结果进行横向地比较;此外,本发明可以同时适用于定量化已发表的公开rna-seq数据和用户自己新测序得到的数据,避免了用户需要掌握和使用不同的软件来处理不同类型数据的困扰,节省用户大量的时间和精力。
附图说明
图1为本发明提供的rna测序数据的基因表达量计算方法流程图。
图2为本发明提供的用户处理公开的rna-seq数据时所需要输入的命令和参数参考示意图。
图3为本发明提供的检测序列的测序质量的参考示意图。
图4为本发明提供的处理用户自己的rna-seq测序数据时所需要输入的命令和参数参考示意图。
图5为本发明提的利用注释数据得到的基因和转录本之间的对应关系参考示意图。
图6为本发明提供的利用stringtie将已定位的序列(bam格式)组装成转录本后得到的gtf格式文件参考示意图。
图7为本发明提供的使用了alignment-based方案得到的基因在基因水平上和转录组水平上的表达量参考示意图。
图8为本发明提供的利用salmon将质量检测合格的或修剪过的序列伪比对到参考转录组后得到的quant.sf文件参考示意图。
图9为本发明提供的使用了alignment-free方案得到的基因在基因水平上和转录组水平上的表达量参考示意图。
具体实施方式
下面对本发明的具体实施方式进行描述,以便于本技术领域的技术人员理解本发明,但应该清楚,本发明不限于具体实施方式的范围,对本技术领域的普通技术人员来讲,只要各种变化在所附的权利要求限定和确定的本发明的精神和范围内,这些变化是显而易见的,一切利用本发明构思的发明创造均在保护之列。
实施例1:
如图1所示,一种rna测序数据的基因表达量计算方法,包括以下步骤:
s1、获取rna测序数据,并对其序列进行质量检测,判断序列质量是否合格;
若是,则进入步骤s3;
若否,则进入步骤s2;
s2、对质量检测不合格的序列进行修剪,进入步骤s3;
s3、根据测序对象的通用名或科学命名,获取对应的参考基因组、参考转录组及注释数据,进入步骤s4;
s4、基于注释数据,提取参考基因组及参考转录组中基因和转录本之间的对应关系;
s5、基于基因和转录本之间的对应关系,计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现测序对象的基因表达量的计算。
本实施例的步骤s1中,rna测序数据包括用户构建的测序数据和公开的测序数据;
当获取的rna测序数据为公开的测序数据时,其被打包压缩成sra格式,需将其转换成fastq格式文本,fastq格式文本保存有测序序列及其测序质量得分信息,用于对公开的测序数据的序列进行质量检测;当获取的rna测序数据为用户构建的测序数据时,直接就是fastq格式文本。
另外,当rna测序数据为公开的测序数据,即rna-seq数据时,需指定获取码,且将获取的rna-seq数据转换成fastq格式文本时,获取对应序列的测序类型,测序类型包括双端序列和单端序列,后续序列修剪及对比和组装等过程都需确定该序列的测序类型。具体的,从sequencereadarchive(sra)下载获取rna-seq数据,下载过程中需要用户指定所下载数据的获取码(accession)如图2所示,这里的获取码可以是来自某个具体的rna-seq数据,也可以是来自某个生物样本(通常包括几个rna-seq数据),或者是来自某个生物项目(通常包括几个生物样本);下载的数据以二分类的形式保存,随后被转换为fastq格式,以文本的形式保存测序序列及其测序质量的得分信息,在数据转换过程中,自动获取序列的测序类型,即双端序列(paired-end)或者单端测序(sing-end);
在本实施例的步骤s1中,对序列进行质量检测包括测序质量检测和测序接头检测,当测序质量检测不合格或接头检测不合格时,序列的质量检测不合格。
进行测序质量检测时,基于测序质量得分信息,当序列中存在测序质量小于设定阈值的碱基或该序列子集的平均测序质量小于设置阈值时,测序质量检测不合格;
进行测序接头检测时,当序列中有超过设定阈值的读段含有测序接头时,测序接头检测不合格。
其中,测序质量小于设定阈值的碱基为低质量的碱基,当设定阈值为20时,每个碱基的测序质量如图3(a)所示,序列子集的平均测序质量小于设置阈值时认为序列子集普遍低质量,当设定阈值为20时,序列子集的质量分数分布如图3(b)所示。进行测序接头检测时,检测的测序接头包括illumina3端接头、illumina5端接头、illuminauniversal接头、nexteratransposasesequence接头和solid接头,对其在序列中出现的频率进行检测,当有超过10%的读段(read)含有测序接头时,则测序接头检测不合格。
本实施例的步骤s2中,对质量检测不合格的序列进行修剪包括去除序列中低于设定阈值的碱基或去除超过设定阈值的测序接头;当rna测序数据为用户构建的测序数据时,还需要在此步骤指定序列的测序类型(如图4所示)。
本实施例的步骤s3中,用户需要指定其研究对象的通用名或者科学命名(图2、图4),然后下载其对应的最新参考基因组、参考转录组和注释数据,下载后的参考基因组数据分散在各个染色体上后被合并为一个整体。
本实施例的步骤s4中,注释数据主要是对基因组的功能进行注释,其中包含了编码基因和其转录组信息,因此能够根据注释数据,提取到基因和转录本之间的对应关系,且通常一个基因对应一个或多个转录本(如图5所示)。
本实施例的步骤s5中,采用依赖参考序列比对(alignment-based)和不依赖参考序列比对(alignment-free)两种方案进行基因表达量的计算,当使用alignment-based方法计算时,上述步骤s5具体为:
a1、基于基因和转录本之间的对应关系,利用hisat2将质量合格的序列或修剪后的序列对比到参考基因组中对其进行定位,将其保存并转化为bam格式;
其中,定位对比后的序列被保存为sam格式,然后被转化为bam格式;
a2、利用stringtie将bam格式的序列组装成转录本,并以gtf格式保存;
其中,gtf格式文本含有转录本数量的间接评估(即coverage覆盖度值,图6倒数第三列)和转录本的起始和终止位置(二者之差为转录本的长度,图6第四和第五列);
a3、基于gtf格式文本及基因和转录本之间的对应关系,利用alignment-based方法计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现基因表达量的计算。
步骤a3中,基于gtf格式文本,质量合格的序列或修剪后的序列在转录组水平上的表达量的计算公式为:
count转录本=coverage*length/read_len
式中,count转录本是转录本的数量,即基因表达量,coverage为转录本的覆盖度,length为转录本的长度,read_len为序列读段的平均长度;
基于基因和转录本之间的对应关系,质量合格的序列或修剪后的序列在基因水平上的表达量的计算公式为:
count基因=∑(count转录本)
式中,count基因为某个基因的表达量,count转录本为基因转录的某个转录本的数量。
将不同测序序列中的基因表达量和转录本表达量进行汇总和合并,将序列的名称作为sample变量(图7)以标注数据的来源。
当采用alignment-free方法进行基因表达量的计算时,上述步骤s5具体为:
b1、基于基因和转录本之间的对应关系,利用salmon将质量合格的序列或修剪后的序列伪对比到参考转录组,将其保存为quant.sf格式纯文本;
其中,quant.sf格式纯文本中含有基因在转录组水平上表达量的评估(即numread值,图8;
b2、基于quant.sf格式纯文本,利用alignment-free方法计算质量合格的序列或修剪后的序列在基因水平上和转录组水平上的表达量,实现基因表达量的计算。
其中,将quant.sf格式纯文本转换为csv格式,得到质量合格的序列或修剪后的序列在转录组水平上的表达量;
所述质量合格的序列或修剪后的序列在基因水平上的表达量的计算公式为:
count基因=∑(count转录本)
式中,count基因为某个基因的表达量,count转录本为基因转录的某个转录本的数量。
将不同测序序列中的基因表达量和转录本表达量进行汇总和合并,将序列的名称作为sample变量(图9)以标注数据的来源。
本发明实施例中提供的alignment-free方案和alignment-based方案得到的结果可能会在具体探测到的表达基因和基因的表达量上有所差异,可以考虑将这两种方案得到的结果进行整合以提高rna-seq分析的准确率(accuracy)和敏感度(sensitivity)。
本发明的有益效果为:
本发明提供的rna测序数据的基因表达量的计算方法,仅需要两个非专业性的参数就可以自动地生成基于alignment-based和alignment-free的基因表达量,可以在提高基因表达量的定量精度的同时方便用户与其他的研究结果进行横向地比较;此外,本发明可以同时适用于定量化已发表的公开rna-seq数据和用户自己新测序得到的数据,避免了用户需要掌握和使用不同的软件来处理不同类型数据的困扰,节省用户大量的时间和精力。
- 还没有人留言评论。精彩留言会获得点赞!