光学活性α-氨基酸的合成方法与流程
本发明涉及使用具有轴手性的N-(2-酰基芳基)-2-[5,7-二氢-6H-二苯并[c,e]氮杂-6-基]乙酰胺化合物作为模板的光学活性α-氨基酸的合成方法。另外,本发明还涉及以用作中间体的N-(2-酰基芳基)-2-[5,7-二氢-6H-二苯并[c,e]氮杂-6-基]乙酰胺化合物的α-氨基酸缩合物作为配体的金属络合物。
背景技术:
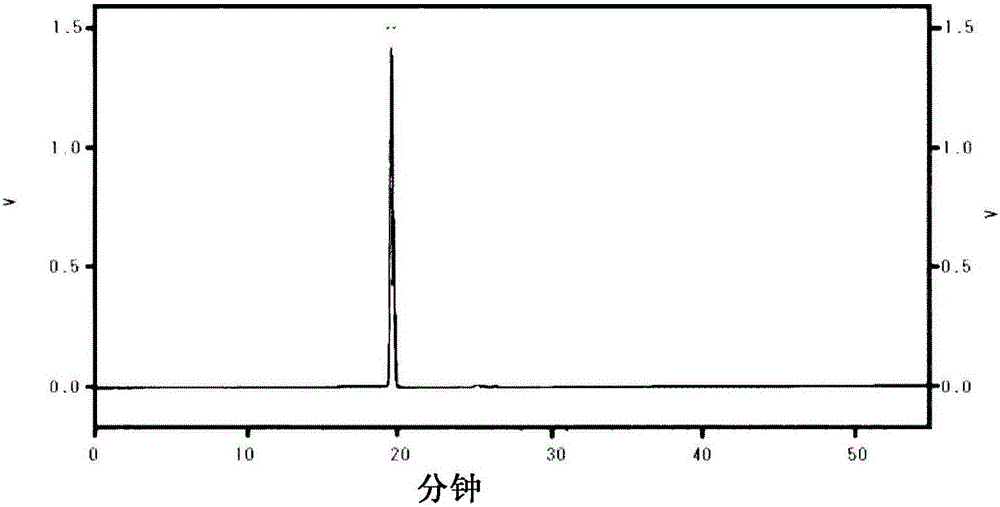
:光学上纯净的α-氨基酸是作为例如各种生理活性物质、设计药品时的结构单元的重要的化合物组。特别是近年来,大量报道了天然中不存在的具有侧链的α-氨基酸本身及掺入有该α-氨基酸的物质具有特异性的生理作用,期望开发出简便地得到光学上纯净的这些α-氨基酸的方法。另外,α-氨基酸的α位为季碳原子的α,α-二取代α-氨基酸具有使肽、蛋白质的高级结构稳定化的效果,并且使肽、蛋白质对水解酶的稳定性提高等,因此,其在药品开发中的重要性日益提高,近年来,其光学活性体的简便获取方法的开发也是首要的课题。作为非天然型的具有侧链的光学活性α-氨基酸的制造方法,传统地报道了利用手性甘氨酸烯醇等价物与各种亲电子剂的非对映选择性烷基化反应、加成反应的方法。例如,在非专利文献1中报道了使用手性双内酰亚胺醚作为手性甘氨酸等价物的方法。但是,该方法中,作为手性甘氨酸等价物的双内酰亚胺醚(bislactimether)的合成需要多个步骤,另外,从酰胺转换为亚胺酯(imidate)的操作困难且需要昂贵的试剂,并且需要将目标氨基酸衍生物与作为手性助剂使用的缬氨酸等氨基酸衍生物分离开来,因此难以应用于大量合成。另外,在非专利文献2中报道了使用咪唑啉酮作为手性甘氨酸等价物的方法。但是,该方法中,作为手性甘氨酸等价物的2-烷基-1,3-咪唑啉酮(2-alkyl-1,3-imidazolidinone)的合成需要多个步骤,而且所生成的咪唑啉酮衍生物是以异构体混合物的形式得到的,因此,需要利用层析等对异构体混合物进行分离。另外,需要昂贵的特戊醛,因此难以应用于大量合成。在非专利文献3中报道了使用5,6-二苯基吗啉-2-酮(5,6-diphenylmorpholin-2-one)作为手性甘氨酸等价物的方法,但该方法中,作为原料的手性1,2-二苯基-2-氨基乙醇(1,2-diphenyl-2-aminoethanol)也昂贵,并且作为手性甘氨酸等价物的5,6-二苯基吗啉-2-酮的合成需要多个步骤,而且在最后步骤中得到氨基酸时,作为手性辅基使用的1,2-二苯基-2-氨基乙醇通常通过还原反应被除去,因此其手性消失,无法回收该手性辅基。因此,该方法也存在成本大的问题,难以应用于大量合成。此外,在非专利文献4报道的方法中,利用使用脯氨酸作为手性源的手性Ni(II)络合物作为手性甘氨酸等价物,并报道了其迈克尔反应,进而还大量报道了该Ni(II)络合物在非对映选择性的烷基化反应、羟醛缩合反应和曼尼希反应中的应用,但脯氨酸的手性中心在立体化学上均不稳定,容易受到差向异构化,难以进行配体的回收和再利用。作为非天然型的具有侧链的光学活性α-氨基酸的制造方法,可以列举以上的4种方法作为使用手性甘氨酸烯醇等价物的显著示例,但每种方法在以千克规模进行工业化的方面均存在缺点,期望开发能够消除这些缺点的新方法。现有技术文献非专利文献非专利文献1:U.Schollkopfetal.,Angew.Chem.Int.Ed.Engl.,1981,20,798.非专利文献2:E.Juaristietal.,J.Org.Chem.,1995,60,6408.非专利文献3:R.M.Williamsetal.,Org.Synth.,2003,80,31非专利文献4:V.A.Soloshonoketal.,J.Am.Chem.Soc.,2005,127,15296技术实现要素:发明所要解决的问题鉴于上述情况,本发明的课题在于提供高收率且高对映选择性地制造各种具有侧链的光学活性α-氨基酸的、能够以千克规模进行工业化的制造方法。另外,本发明的课题还在于提供光学活性α,α-二取代α-氨基酸的简便制造方法。此外,本发明的课题还在于提供在上述光学活性α-氨基酸和光学活性α,α-二取代α-氨基酸的制造方法中有用的中间体。用于解决问题的方法即,本发明涉及以下内容。[1]一种光学活性α-氨基酸或其盐的制造方法,其特征在于,通过烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应在下述式(1)所示的金属络合物的α-氨基酸部分结构的α碳上引入取代基,然后,利用酸分解使α-氨基酸或其盐游离,(式中,R1表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、卤素原子或硝基,R2表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基或可以被取代的杂芳基,R3、R4各自独立地表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基或卤素原子,两个R3可以相同也可以不同,两个R4可以相同也可以不同,R3和R4可以与它们所键合的碳原子一同形成环,R5表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的羧基(-CO2R7)或卤素原子,两个R5可以相同也可以不同,R6表示氢、可以被取代的烷基、可以被取代的环烷基或卤素原子,两个R6可以相同也可以不同,两个R6可以与它们所键合的碳原子一同形成环,R7表示氢、可以被取代的烷基、可以被取代的芳基或可以被取代的杂芳基,R8表示氢、可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基或可以被取代的杂芳基烷基,*表示手性轴。M表示二价金属阳离子)。[2]如上述[1]所述的方法,其中,所述式(1)的金属络合物是R8为氢的下述式(1-1)的金属络合物,(式中,R1~R6、*和M与上述[1]的R1~R6、*和M含义相同)。[3]如上述[1]或[2]所述的方法,其中,所述式(1)的金属络合物是R1为氢、氯、甲基或硝基,在两组R3和R4中的任意一组中,R3和R4均与所键合的芳香环的碳原子一同进一步形成芳香环或脂环式结构,R5和R6均为氢,M为镍、铜、钯或铂阳离子的下述式(1b)的金属络合物,(式中,R2、R8和*与上述[1]的R2、R8和*含义相同)。[4]如上述[1]~[3]中任一项所述的制造方法,其中,在所述α碳上引入取代基之后,在酸分解之前,进一步包括提高α碳的光学纯度的步骤。[5]如上述[4]所述的方法,其中,所述提高光学纯度的步骤为在碱性条件下加热的步骤。[6]如上述[1]~[5]中任一项所述的制造方法,其中,所述光学活性α-氨基酸或其盐为非天然氨基酸或其盐。[7]一种二取代体的光学活性α-氨基酸或其盐的制造方法,其特征在于,通过烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应在上述[2]所述的式(1-1)所示的金属络合物的α-氨基酸部分结构的α碳上引入一个取代基而形成下述式(1-2)所示的金属络合物,接着,进一步进行烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应,由此在所述α碳上引入另一个取代基,(式中,R1~R6、*和M与上述[1]的R1~R6、*和M含义相同。R9表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基或可以被取代的芳氧基羰基)。[8]一种式(1-1)所示的金属络合物,(式中,R1~R6、*和M与上述[1]的R1~R6、*和M含义相同)。[9]一种式(2)所示的金属络合物,(式中,R1~R6、*和M与上述[1]的R1~R6、*和M含义相同,R12和R13各自独立地表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基、可以被取代的芳氧基羰基或卤素原子)。[10]一种式(2)所示的金属络合物,(式中,R1~R6、*和M与上述[1]的R1~R6、*和M含义相同,R12和R13各自独立地表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基、可以被取代的芳氧基羰基或卤素原子,或者,R12和R13可以与它们所键合的碳原子一同形成环)。[11]如上述[8]~[10]中任一项所述的金属络合物,其中,R1为氢、氯、甲基或硝基,在两组R3和R4中的任意一组中,R3和R4均与所键合的芳香环的碳原子一同进一步形成芳香环或脂环式结构,R5和R6均为氢,R2为式(1-1a)所示的芳基,(R14表示氢原子或卤素原子)。发明效果根据本发明,能够高收率且高对映选择性地以千克规模制造具有所期望的手性的光学活性α-氨基酸。另外,根据本发明,还能够高收率且高对映选择性地并且简便地制造在药品开发等中的重要性日益提高的光学活性α,α-二取代α-氨基酸。附图说明图1是实施例1-2中制备的化合物的HPLC色谱图。图2是实施例1-3中制备的化合物的HPLC色谱图。图3是实施例2-1中制备的化合物的HPLC色谱图。图4是实施例2-2中制备的化合物的HPLC色谱图。图5是实施例2-3中制备的化合物的HPLC色谱图。图6是实施例2-4中制备的化合物的HPLC色谱图。图7是实施例2-5中制备的化合物的HPLC色谱图。图8是实施例2-6中制备的化合物的HPLC色谱图。图9是实施例2-7中制备的化合物的HPLC色谱图。图10是实施例2-8中制备的化合物的HPLC色谱图。图11是实施例2-9中制备的化合物的HPLC色谱图。图12是实施例3-1中制备的化合物的HPLC色谱图。图13是实施例3-2中制备的化合物的HPLC色谱图。图14是实施例3-3中制备的化合物的HPLC色谱图。图15是实施例3-4中制备的化合物的HPLC色谱图。图16是实施例4-2中制备的化合物的HPLC色谱图。图17是实施例5-1中制备的化合物的HPLC色谱图。图18是实施例5-2中制备的化合物的HPLC色谱图。图19是实施例5-3中制备的化合物的HPLC色谱图。图20是实施例5-4中制备的化合物的HPLC色谱图。图21是实施例5-5中制备的化合物的HPLC色谱图。图22是实施例6中制备的化合物的HPLC色谱图。图23是实施例7-1中制备的化合物的HPLC色谱图。图24是实施例7-2中制备的化合物的HPLC色谱图。图25是实施例8中制备的化合物的HPLC色谱图。图26是实施例9-1中制备的化合物的HPLC色谱图。图27是实施例9-2中制备的化合物的HPLC色谱图。图28是参考例2中制备的化合物的HPLC色谱图。具体实施方式本发明涉及利用通式(3)的光学活性N-(2-酰基芳基)-2-[5,7-二氢-6H-二苯并[c,e]氮杂-6-基]乙酰胺化合物的立体结构来制造具有所期望的手性的光学活性α-氨基酸和α,α-二取代α-氨基酸的方法。(式中,R1~R6和*与上述[1]的R1~R6和*含义相同)。本发明所涉及的化学反应的概要如下所述。在通式(1)所示的金属络合物的α-氨基酸部分结构的α碳上引入侧链,然后,利用酸分解使α-氨基酸游离。(式中,R1表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、卤素原子或硝基,R2表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基或可以被取代的杂芳基,R3、R4各自独立地表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基或卤素原子,两个R3可以相同也可以不同,两个R4可以相同也可以不同,R3和R4可以与它们所键合的碳原子一同形成环,R5表示氢、可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的羧基(-CO2R7)或卤素原子,两个R5可以相同也可以不同,R6表示氢、可以被取代的烷基、可以被取代的环烷基或卤素原子,两个R6可以相同也可以不同,两个R6可以与它们所键合的碳原子一同形成环,R7表示氢、可以被取代的烷基、可以被取代的芳基或可以被取代的杂芳基,R8表示氢、可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基或可以被取代的杂芳基烷基,*表示手性轴。M表示二价金属阳离子)。另外,本发明所涉及的反应还包含通过对通式(2)所示的金属络合物进行酸分解而分离出α碳为季碳原子的α-氨基酸的反应。(式中,R1~R6、*和M与上述式(1)的R1~R6、*和M含义相同,R12和R13各自独立地表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基、可以被取代的芳氧基羰基或卤素原子,或者,R12和R13可以与它们所键合的碳原子一同形成环)。需要说明的是,上述R12可以与后述的R9或R11含义相同,上述R13可以与后述的R10含义相同。即,式(2)所示的化合物包含后述的式(2-1)所示的化合物和式(2-1’)所示的化合物。在本发明的一个方式中,[A-1]使通式(9)的金属盐MXn与通过通式(3)的化合物与式(4)的甘氨酸的缩合反应生成的亚胺化合物反应,形成通式(1-1)的金属络合物,[A-2]在上述通式(1-1)的金属络合物的α-氨基酸部分结构的α碳上,通过烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应等与亲电子剂的反应等引入侧链,得到通式(1-2)的金属络合物后,[A-3]对上述通式(1-2)的金属络合物进行酸分解,由此高对映选择性地制造通式(6)的光学活性α-氨基酸。另外,在本发明的另一个方式中,[B-1]在上述步骤[A-2]之后,通过烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应等在α-氨基酸部分结构的α碳上再引入另一个侧链,得到通式(2-1)的金属络合物后,[B-2]对上述通式(2-1)的金属络合物进行酸分解,由此高对映选择性地制造通式(7)的光学活性α,α-二取代α-氨基酸。在本发明的又一个方式中,[C-1]使通式(9)的金属盐MXn与通过通式(3)的化合物与通式(5)的α-氨基酸的缩合反应生成的亚胺化合物反应,形成通式(1-1’)的金属络合物,[C-2]在上述通式(1-1’)的金属络合物的α-氨基酸部分结构的α碳上,通过烷基化反应、羟醛缩合反应、迈克尔反应或曼尼希反应等引入侧链,得到通式(2-1’)的金属络合物后,[C-3]对上述通式(2-1’)的金属络合物进行酸分解,由此高对映选择性地制造通式(8)的光学活性α,α-二取代α-氨基酸。需要说明的是,在上述步骤[A-2]之后和上述步骤[C-1]之后,可以包括对所得到的化合物进行加热的步骤。如后所述,通过加热,能够使α-氨基酸部分结构的α碳的立体构型与该络合物中的手性轴的立体构型相应地转换为S或R构型中的任意一种构型。本发明所涉及的化学反应的整体线路图(上述步骤[A-1]~[A-3]、[B-1]~[B-2]、[C-1]~[C-3])如下所示。通式(3)的化合物存在式(3-S体)和式(3-R体)所示的两种光学异构体。本发明的光学活性α-氨基酸的制造方法中,利用通式(3)的立体结构来控制最终得到的光学活性α-氨基酸的α碳的立体构型。即,本发明包括通过适当选择地使用式(3-S体)和式(3-R体)的光学异构体来制造具有所期望的手性、光学上纯净的或光学纯度高的光学活性α-氨基酸的方法。以下,对本发明进行详细说明。首先,对式(3)所示的化合物进行说明。(式中,R1、R2、R3、R4、R5、R6和*与上述式(1)含义相同)。R1所示的可以被取代的烷基中的“烷基”没有特别限定,可以为直链状,也可以为支链状。作为上述“烷基”,可以列举例如碳原子数1~20的烷基等,具体而言,可以列举甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基、十二烷基、十五烷基、十六烷基、十八烷基等。R1所示的可以被取代的炔基中的“炔基”没有特别限定。作为上述“炔基”,可以列举例如碳原子数2~20的炔基、优选列举碳原子数2~10的炔基等,具体而言,可以列举乙炔基、丙炔基等。R1所示的可以被取代的烯基中的“烯基”没有特别限定。作为上述“烯基”,可以列举例如碳原子数2~20的烯基、优选列举碳原子数2~10的烯基等,具体而言,可以列举乙烯基、烯丙基、丁烯基、己烯基等。R1所示的可以被取代的烷氧基中的“烷氧基”没有特别限定。作为上述“烷氧基”,可以列举例如碳原子数1~20的烷氧基、优选列举碳原子数1~10的烷氧基等,具体而言,可以列举甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔丁氧基、戊氧基等。R1所示的可以被取代的环烷基中的“环烷基”没有特别限定。作为上述“环烷基”,可以列举例如碳原子数3~12的环烷基、优选列举碳原子数3~10的环烷基等,具体而言,可以列举环丙基、环丁基、环戊基、环己基、环庚基等。R1所示的可以被取代的芳基中的“芳基”没有特别限定。作为上述“芳基”,可以列举例如碳原子数6~20的芳基等,具体而言,可以列举苯基、1-萘基、2-萘基、蒽基、菲基、2-联苯基、3-联苯基、4-联苯基、三联苯基等。R1所示的可以被取代的杂芳基中的“杂芳基”没有特别限定。作为上述“杂芳基”,可以列举例如优选含有1~3个选自氮原子、硫原子和氧原子等中的原子作为杂原子的杂芳基,具体而言,可以列举呋喃基、噻吩基、唑基、异唑基、噻唑基、异噻唑基、吡咯基、咪唑基、吡唑基、吡啶基、嘧啶基、哌啶基、酞嗪基、三嗪基、吲哚基、异吲哚基、喹啉基、异喹啉基、苯并呋喃基、二苯并呋喃基等。R1所示的卤素原子没有特别限定。作为上述卤素原子,可以列举例如氟原子、氯原子、溴原子、碘原子等。作为R1中的“取代基”,没有特别限定。作为上述“取代基”,可以列举例如烷基(例如,甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基等)、炔基(例如,乙炔基、丙炔基等)、烯基(例如乙烯基、烯丙基、丁烯基、己烯基等)、烷氧基(例如,甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔丁氧基、戊氧基等)、环烷基(例如,环丙基、环丁基、环戊基、环己基、环庚基等)、芳基(例如,苯基、1-萘基、2-萘基、蒽基、菲基、2-联苯基、3-联苯基、4-联苯基、三联苯基等)、杂芳基(例如,呋喃基、噻吩基、唑基、异唑基、噻唑基、异噻唑基、吡咯基、咪唑基、吡唑基、吡啶基、嘧啶基、哌啶基、酞嗪基、三嗪基、吲哚基、异吲哚基、喹啉基、异喹啉基、苯并呋喃基、二苯并呋喃基等)、芳烷基(例如,苄基、苯乙基、苯丙基、萘甲基等)、卤代烷基(例如,三氟甲基、三氯甲基等)、卤代烷氧基(例如,氟甲氧基、二氟甲氧基、三氟甲氧基、三氟乙氧基、四氟乙氧基等)、卤素原子(例如,氟原子、氯原子、溴原子、碘原子等)、羟基、被保护的羟基(作为羟基的保护基,例如为乙酰基、苯甲酰基、甲氧基甲基、四氢吡喃基、三甲基甲硅烷基、叔丁基二甲基甲硅烷基、碳酸酯基等)、氨基、被保护的氨基(作为氨基的保护基,例如为甲酰基、乙酰基、苯甲酰基、苄氧基羰基、邻苯二甲酰基、氨基甲酰基、脲基等)、芳基氨基、杂芳基氨基、巯基、硝基、腈基、羧基、烷氧基羰基等。这些取代基的碳原子数没有特别限定,优选为1~10。上述“取代基”的数量没有特别限定。上述“取代基”的数量例如可以为1~4个,优选为1~2个,更优选为1个。R1的键合位置没有特别限定。R1的键合位置可以为3位、4位、5位、6位中的任意一个位置,优选为4位。作为R2所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基或可以被取代的杂芳基,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。作为R3、R4所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基或卤素原子,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。作为R3和R4与它们所键合的碳原子一同形成的环,没有特别限定,可以为脂环式环,也可以为芳香环。作为上述环,可以列举例如环烷烃环、环烯烃环、芳基环、杂芳基环等,具体而言,可以列举环戊烷、环己烷、环戊烯、环己烯、苯环、萘环、吡啶环等。上述环的碳原子数没有特别限定,优选为3~15。作为R5所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的烷氧基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、卤素原子,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。作为R6所示的可以被取代的烷基、可以被取代的环烷基或卤素原子,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。R1优选为氢、氯、甲基或硝基。R2优选为可以被取代的芳基,更优选为苯基或被卤素原子取代的苯基。两个R3优选相同。另外,两个R4优选相同。另外,更优选如下述式(3-1)所示R3和R4与它们所键合的碳原子一同形成环。(式中,R1、R2、R5、R6和*与上述含义相同)。另外,两个R5优选相同,更优选两个R5均为氢。两个R6优选相同,更优选两个R6均为氢。此外,上述式(3-1)优选为式(3-1a)所示的化合物。(式中,R1、R3、R4、R5、R6和*与上述含义相同,R14表示氢原子或卤素原子)。作为式(3)所示的化合物或其盐,可以列举例如以下的结构式(3-1-1)~(3-1-7)所示的化合物或其盐等。接着,对式(3)所示的化合物的制造方法进行说明。例如,可以通过使式(3-a)所示的化合物或其盐、式(3-b)所示的化合物或其盐与式(3-c)所示的化合物或其盐反应来制造。(式中,R1和R2与上述含义相同)(式中,R6与上述含义相同,L1和L2各自独立地表示离去基团)(式中,R3、R4、R5和*所示的符号与上述含义相同)。上述式(3)所示的化合物的制造方法中,使式(3-a)、式(3-b)和式(3-c)所示的化合物反应的方法依照公知方法、自身公知的方法或基于该方法的方法来进行即可。例如,可以通过使式(3-a)所示的化合物与式(3-b)所示的化合物反应,使所得到的反应产物进一步与式(3-c)所示的化合物反应来制造。式(3-a)所示的化合物或其盐可以通过公知的方法来制造,也可以使用市售品。式(3-a)所示的化合物或其盐优选为式(3-a-1)所示的化合物。(式中,R1和与上述含义相同,R14表示氢原子或卤素原子)。式(3-a-1)所示的化合物或其盐中,作为R1,可以列举例如式(3)中所例示的基团等。另外,作为卤素,可以列举例如式(3)中所例示的卤素等。式(3-b)所示的化合物或其盐中,L1和L2各自独立地表示离去基团,R6与上述含义相同。作为离去基团,只要是通常作为离去基团所公知的基团则没有特别限定,可以列举例如卤素原子、甲苯磺酸基(OTs)或甲磺酸基(OMs)等。L1和L2优选为相同的基团,更优选均为卤素原子。作为卤素原子,更优选为氯原子或溴原子。作为式(3-b)所示的化合物,可以列举例如ClCH2COCl、BrCH2COBr等。式(3-b)所示的化合物或其盐可以通过公知的方法来制造。作为从式(3-b)所示的化合物衍生的乙酰苯胺化合物,可以使用例如文献(T.K.Ellisetal.,J.Org.Chem.,2006,71,8572-8578.)中记载的物质等。接着,对上述式(3-c)所示的化合物进行说明。式(3-c)的化合物优选为式(3-c-1)所示的化合物。(式中,R5和*所示的符号与上述含义相同)。作为式(3-c-1)所示的化合物,可以使用例如文献(N.Maigrotetal.,J.Org.Chem.,1985,50,3916-3918.)中记载的物质等。接着,对上述式(3)的制造中的使式(3-a)、式(3-b)和式(3-c)所示的化合物反应的条件进行说明。关于式(3-b)所示的化合物或其盐的使用量,只要使反应进行则没有特别限定,具体而言,相对于式(3-a)所示的化合物或其盐1摩尔,通常可以为约0.5摩尔~约10摩尔,优选为约1.0摩尔~约3.0摩尔。关于式(3-c)所示的化合物或其盐的使用量,只要使反应进行则没有特别限定,具体而言,相对于式(3-a)所示的化合物或其盐1摩尔,通常可以为约0.5摩尔~约5.0摩尔,优选为约0.5摩尔~约2.0摩尔。式(3)所示的化合物或其盐的上述制造方法中,作为反应中使用的溶剂,没有特别限定,可以列举例如醇类(甲醇、乙醇、异丙醇、叔丁醇等)、醚类(乙醚、四氢呋喃(THF)、1,4-二氧杂环己烷、1,2-二甲氧基乙烷等)、卤代烃类(二氯甲烷、氯仿、1,2-二氯乙烷、四氯化碳等)、芳香族烃类(苯、甲苯、二甲苯、吡啶等)、脂肪族烃类(己烷、戊烷、环己烷等)、腈类(乙腈、丙腈等)、酰胺类(N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、N-甲基吡咯烷酮)等有机溶剂,其中,从反应效率的观点出发,更优选乙腈、丙腈等腈类、N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、N-甲基吡咯烷酮等酰胺类。式(3)所示的化合物或其盐的上述制造方法中,作为反应中使用的碱,没有特别限定,可以列举例如氢氧化钾、氢氧化钠、氢氧化锂、碳酸氢钠、碳酸钾、碳酸钠、碳酸铯、乙酸钠、乙酸钾、乙酸锂、硝酸铷、硝酸锂、亚硝酸铷、亚硫酸钠、氰酸钠、氰酸锂、硫氰酸钠、硫氰酸钾、硬脂酸钠、硬脂酸铯、硼氢化钠、硼氢化钾、硼氢化锂、四苯硼钠(フェニル化ホウ素ナトリウム)、苯甲酸钠、苯甲酸锂等,其中,从反应效率的观点出发,优选氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯等。式(3)所示的化合物或其盐的上述制造方法中,为了得到光学上纯净的目标物质,可以进一步实施分离、纯化步骤。分离、纯化方法没有特别限定,可以采用本领域中通常使用的各种方法。作为分离方法,可以具体列举浓缩、萃取、过滤、洗涤等;作为纯化方法,可以具体列举例如结晶化法(再结晶或悬浊等)、选择性溶解法、使用光学异构体分离用柱等的物理光学拆分等。作为上述再结晶,可以列举与非手性酸(盐酸、硫酸、甲磺酸、甲酸、三氟乙酸等)形成盐后使其再结晶的方法或者使用手性酸(扁桃酸、酒石酸、二苯甲酰基酒石酸、二甲苯酰基酒石酸、10-樟脑磺酸、苹果酸)的非对映异构体盐法等。式(3)所示的化合物或其盐的制造中的更具体的反应条件可以参考后述的实施例。本发明的优选方式中,使用上述式(3)所示的化合物,制造下述式(1)。(式中,R1、R2、R3、R4、R5、R6和*所示的符号与上述含义相同,R8表示氢、可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基或可以被取代的杂芳基烷基,M表示二价金属阳离子。需要说明的是,作为R1~R6的具体例,可以列举例如式(3)中所例示的基团等)。式(1)所示的金属络合物中,作为M所示的上述二价金属阳离子,没有特别限定,可以列举例如镁、钙、锶、钡等碱土金属阳离子、镉、钛、锆、镍(II)、钯、铂、锌、铜(II)、汞(II)、铁(II)、钴(II)、锡(II)、铅(II)、锰(II)等过渡金属阳离子等。其中,优选为镍、铜、钯或铂的阳离子。式(1)所示的金属络合物中,*所示的联苯基的部分结构具有轴手性。式(1)所示的金属络合物优选为R3和R4与它们所键合的碳原子一同形成了芳香环或脂环式结构的、式(1a)所示的金属络合物。(式中,R1、R2、R5、R6、R8、*和M与上述含义相同)。进一步,上述式(1a)的金属络合物优选为R5和R6均为氢、M为镍、铜、钯或铂阳离子的式(1b)所示的金属络合物。(式中,R1、R2、R3、R4、R8和*与式(1a)含义相同,M为镍、铜、钯或铂阳离子)。另外,上述式(1)所示的金属络合物包含式(1-1)所示的金属络合物和式(1-1’)所示的金属络合物。(式中,R1、R2、R3、R4、R5、R6、*和M所示的符号与上述含义相同)。(式中,R1、R2、R3、R4、R5、R6、*和M所示的符号与上述含义相同,R11可以与除氢原子以外的上述R8相同。**表示手性碳原子)。本发明的特别优选的方式中,上述式(1-1)或式(1-1’)所示的金属络合物为下述金属络合物:R1为氢、氯、甲基或硝基,在两组R3和R4中的任意一组中,R3和R4均与所键合的芳香环的碳原子一同进一步形成芳香环或脂环式结构,R5和R6均为氢,R2为式(1-1a)所示的芳基。(R14表示氢原子或卤素原子)。接着,对上述式(1-1)所示的金属络合物的制造方法进行说明。该步骤相当于上述整体线路图的步骤[A-1]。使式(3)、式(4)所示的甘氨酸或其盐和式(9)所示的金属化合物在碱的存在下反应,可以得到式(1-1)所示的金属络合物。(式中,R1、R2、R3、R4、R5、R6和*与上述含义相同)。MXn(9)(式中,M表示二价金属阳离子,X表示一价或二价阴离子,在X为一价阴离子的情况下,n=2,在X为二价阴离子的情况下,n=1)。式(1-1)所示的金属络合物的制造方法中,作为反应中使用的溶剂,优选醇类(甲醇、乙醇、异丙醇、叔丁醇、异丁醇等)。关于溶剂的使用量,只要使反应进行则没有特别限定,相对于式(3)所示的化合物或其盐1重量份,通常可以设定为约1.0倍容量~约150倍容量,从制造效率的观点出发,优选设定为约5倍容量~约50倍容量。式(4)所示的甘氨酸或其盐的使用量没有特别限定,相对于式(3)所示的化合物或其盐1摩尔,通常可以设定为约0.1摩尔~约10摩尔,从反应效率的观点出发,优选为约0.3摩尔~约5摩尔。式(9)所示的金属化合物的使用量没有特别限定,例如,相对于式(3)所示的化合物或其盐1摩尔,通常可以设定为约0.1摩尔~约10摩尔,从反应效率的观点出发,优选为约0.5摩尔~约8.0摩尔。作为式(1-1)所示的金属络合物的制造方法中使用的碱,没有特别限定,可以与例如式(3)所示的化合物或其盐的制造方法中使用的碱相同,其中,从反应效率的观点出发,优选碳酸钾、碳酸钠、碳酸铯、氢氧化钾、氢氧化钠、氢氧化锂。作为上述碱的使用量,没有特别限定,相对于式(3)所示的化合物1摩尔,通常可以设定为约0.1摩尔~约20摩尔,从反应效率的观点出发,优选为0.5~10摩尔。上述制造方法中,只要使反应充分进行,则反应时间没有特别限定,通常可以设定为0.1~72小时,从制造效率的观点出发,优选为0.1~48小时,特别优选为0.1~20小时。另外,进行上述反应时的压力没有特别限定,可以在常压下、加压下、减压下的任意一种条件下进行。具体而言,例如,通常可以设定为约0.1个大气压~约10个大气压。关于进行上述反应时的反应温度,只要使反应进行则没有特别限定,例如,通常可以设定为0℃~100℃,从反应效率的观点出发,优选为0℃~80℃、更优选为5℃~60℃。这样得到的式(1-1)所示的金属络合物可以用于下一步骤(即,整体线路图中所示的步骤[A-2])。接着,对上述式(1-1’)所示的金属络合物的制造方法进行说明。该步骤相当于上述整体线路图的[C-1]所示的步骤。使式(3)、式(5)所示的光学活性α-氨基酸(或任意比例的混合物)或其盐以及式(9)所示的金属化合物在碱的存在下反应,可以得到式(1-1’)所示的金属络合物。(式中,R1、R2、R3、R4、R5、R6和*与上述含义相同)。(式中,R11与上述式(1-1’)含义相同)。MXn(9)(式中,M、X、n与上述含义相同)。作为上述式(5)所示的光学活性α-氨基酸或其盐,可以列举例如丙氨酸(Ala)、精氨酸(Arg)、天冬酰胺(Asn)、天冬氨酸(Asp)、半胱氨酸(Cys)、谷氨酰胺(Gln)、谷氨酸(Glu)、组氨酸(His)、异亮氨酸(Ile)、亮氨酸(Leu)、赖氨酸(Lys)、蛋氨酸(Met)、苯丙氨酸(Phe)、丝氨酸(Ser)、苏氨酸(Thr)、色氨酸(Trp)、酪氨酸(Tyr)、缬氨酸(Val)等α-氨基酸及它们的盐等,另外,可以列举非天然型的光学活性α-氨基酸及它们的盐等。这些α-氨基酸或其盐可以为L型、D型或它们的任意比例的混合物。式(1-1’)所示的金属络合物的制造中使用的溶剂的种类和使用量、上述式(5)所示的α-氨基酸或其盐的使用量、上述式(9)所示的金属化合物的使用量、使用的碱的种类和使用量、反应时间、反应时的压力、反应温度可以与上述式(1-1)所示的金属络合物的制造方法相同。这样得到的式(1-1’)所示的金属络合物具有手性碳原子(**所示的、α-氨基酸部分结构的α碳原子)。(式中,R1、R2、R3、R4、R5、R6、R11、*、M和**所示的符号与上述含义相同)。接着,对作为本发明的一个方式的、使用式(1-1)所示的金属络合物并且高收率且高对映选择性地制造光学活性α-氨基酸的方法进行具体说明。该步骤相当于上述整体线路图的步骤[A-2]和[A-3]。首先,对上述步骤[A-2]进行说明。该步骤为在式(1-1)所示的金属络合物的α-氨基酸部分结构的α碳上引入侧链而得到式(1-2)所示的金属络合物的步骤。(式中,R1、R2、R3、R4、R5、R6、*、M和**所示的符号与上述式(1-1’)含义相同,R9表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基或可以被取代的芳氧基羰基)。作为R9所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基或可以被取代的杂芳基,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。另外,作为R9所示的可以被取代的芳烷基,可以列举可以被取代的苄基、苯乙基、苯丙基、萘甲基或萘乙基等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。另外,作为R9所示的可以被取代的杂芳基烷基,可以列举由可以被取代的杂芳基和可以被取代的烷基构成的基团。作为上述可以被取代的杂芳基和可以被取代的烷基,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。另外,作为R9所示的可以被取代的烷氧基羰基,可以列举由羰基和可以被取代的烷氧基构成的基团。作为上述可以被取代的烷氧基,可以列举由可以被取代的烷基和氧原子构成的基团,可以列举例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔丁氧基、戊氧基、癸氧基、环戊氧基、环己氧基、薄荷基氧基(メンチルオキシ基)、氯甲氧基、氟甲氧基、三氟甲氧基、甲氧基甲氧基、乙氧基甲氧基、甲氧基乙氧基、苄氧基、4-氯苄氧基、4-甲基苄氧基、4-甲氧基苄氧基、3-苯氧基苄氧基等。作为上述可以被取代的烷基,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。此外,作为R9所示的可以被取代的芳氧基羰基,可以列举由羰基和可以被取代的芳氧基构成的取代基,可以列举例如苯氧基羰基、2-甲基苯氧基羰基、4-甲基苯氧基羰基、4-甲氧基苯氧基羰基、萘氧基羰基等。作为上述可以被取代的芳氧基,可以列举由可以被取代的芳基和氧原子构成的基团,可以列举例如苯氧基、2-甲基苯氧基、4-氯苯氧基、4-甲基苯氧基、4-甲氧基苯氧基、3-苯氧基苯氧基等。作为上述可以被取代的芳基,可以列举例如R1中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。上述在α碳上引入侧链的方法没有特别限定,可以采用例如烷基化反应、羟醛缩合反应、迈克尔反应、曼尼希反应等与亲电子剂的反应等。需要说明的是,烷基化反应、羟醛缩合反应、迈克尔反应和曼尼希反应可以依照公知方法、自身公知的方法或基于这些方法的方法来进行。对上述烷基化反应进行说明。上述步骤[A-2]中的上述烷基化反应是使式(1-1)的金属络合物与烷基化剂(亲电子剂)在碱的存在下反应而在式(1-1)的金属络合物的α-氨基酸部分结构的α碳上引入烷基的反应。作为上述烷基化剂,可以列举例如卤代烷、硫酸酯、芳香族磺酸酯、草酸酯、羧酸酯、磷酸酯、原酸酯、二甲基甲酰胺缩醛、三氟甲磺酸酯、烷基铵盐、烷基重氮盐、烷基氧盐、烷基锍盐、烷基碘盐、氟硫酸酯、碳酸二烷基酯、氯甲酸酯、氰基甲酸酯(例如Mander试剂)等。其中,特别优选使用卤代烷、氰基甲酸酯。作为上述碱,只要使反应进行则没有特别限定,可以列举例如甲醇钠等碱金属的醇盐、氨基钠等碱金属的酰胺等,从在所使用的反应溶剂中的碱性强的观点出发,优选甲醇钠。上述碱的添加量没有特别限定,相对于式(1-1)的金属络合物1摩尔,通常为1~20摩尔,从反应效率的观点出发,优选为1.5~5摩尔。作为烷基化反应中使用的溶剂,没有特别限定,可以列举例如丙酮、甲苯、苯、二甲亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、二甲氧基乙烷(DME)、乙醚、四氢呋喃(THF)、二氧杂环己烷、乙腈、二氯甲烷等。关于反应温度,只要使反应进行则没有特别限定,例如通常为-20℃~25℃,从反应效率的观点出发,优选为-10℃~10℃,更优选为-5℃~5℃。关于反应时间,只要使反应进行则没有特别限定,例如通常为0.1小时~30小时,从反应效率的观点出发,优选为0.1~24小时。关于本发明的烷基化反应中使用的烷基化剂的量,只要使反应进行则没有特别限定,相对于式(1-1)的金属络合物1摩尔,例如通常可以设定为0.5~5摩尔,从反应效率的观点出发,优选为1~5摩尔。上述烷基化反应依赖于式(1-1)的金属络合物的手性轴的立体构型而高收率且高对映选择性地进行。即,在式(1-1)的金属络合物的手性轴的立体构型为S构型的情况下,通过烷基化反应引入侧链时,α-氨基酸部分结构的立体构型成为D型,在式(1-1)的金属络合物的手性轴的立体构型为R构型的情况下,α-氨基酸部分结构的立体构型成为L型。(式中,R1、R2、R3、R4、R5、R6和M所示的符号与式(1-1)含义相同,斜体字的S或R表示手性轴的立体构型,R9与上述含义相同)。接着,对上述羟醛缩合反应进行说明。上述步骤[A-2]中的上述羟醛缩合反应是通过式(1-1)的金属络合物与芳香族醛或脂肪族醛(亲电子剂)的反应而在β碳上引入具有羟基的侧链的反应。本发明中,上述芳香族醛和脂肪族醛没有特别限定,可以列举例如后述的在R15上键合有醛基的化合物等。上述羟醛缩合反应中,可以使用酸催化剂、碱催化剂中的任意一种催化剂。从反应效率的观点出发,优选碱催化剂。作为上述碱催化剂,没有特别限定,可以列举例如二氮杂双环十一碳烯(DBU)、二氮杂双环壬烯(DBN)、三氮杂双环癸烯(TBD)、二氮杂双环[2.2.2]辛烷(DABCO)、三乙胺、二异丙基乙胺、吡啶、4-二甲基氨基吡啶、甲醇钠等碱金属的醇盐、叔丁醇钾、氢化钠、丁基锂、二异丙基氨基锂、双(三甲基甲硅烷基)胺基锂等,其中,优选二氮杂双环十一碳烯(DBU)、二氮杂双环壬烯(DBN)等。关于催化剂的添加量,只要使反应进行则没有特别限定,相对于作为底物的式(1-1)的化合物1摩尔量,通常为1~6摩尔,从反应效率的观点出发,优选为2~5摩尔,更优选为3摩尔。羟醛缩合反应中使用的溶剂没有特别限定,可以列举甲醇、乙醇、异丙醇、叔丁醇等醇类、二甲亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、四氢呋喃(THF)、二氧杂环己烷、乙腈、二氯甲烷等。反应温度没有特别限定,通常为-40℃~40℃,从反应效率的观点出发,优选为-20℃~20℃,更优选为-10℃~0℃。反应时间没有特别限定,通常可以设定为0.1小时~30小时,从反应效率的观点出发,优选为0.1~2小时。关于反应中使用的芳香族醛和脂肪族醛的量,只要使反应进行则没有特别限定,例如,相对于式(1-1)的金属络合物1摩尔,通常为0.5~10摩尔,从反应效率的观点出发,优选为1~8摩尔,更优选为2~7摩尔。上述羟醛缩合反应依赖于式(1-1)的金属络合物的手性轴的立体构型而高收率且高对映选择性地进行,选择性地或优先地提供单一的立体异构体。即,在式(1-1)的金属络合物的手性轴的立体构型为S构型的情况下,通过羟醛缩合反应引入侧链时,α-氨基酸部分结构的α碳的立体构型成为S构型且β碳的立体构型成为R构型,在式(1-1)的金属络合物的手性轴的立体构型为R构型的情况下,α-氨基酸部分结构的立体构型成为R构型且β碳的立体构型成为S构型。(式中,R1、R2、R3、R4、R5、R6和M所示的符号与式(1-1)含义相同,斜体字的S或R表示立体构型,R15表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基或可以被取代的杂芳基烷基)。作为R15所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基,可以列举例如R9中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。接着,对迈克尔反应进行说明。上述步骤[A-2]中的上述迈克尔反应是使式(1-1)的金属络合物与各种迈克尔反应受体在碱的存在下反应而在α-氨基酸部分结构的α碳上引入侧链的反应。作为反应中使用的上述碱,没有特别限定,可以列举例如氢氧化钠、氢氧化锂、碳酸氢钠、碳酸钠、碳酸钾、碳酸铯、乙酸钠、乙酸钾、乙酸锂、硝酸铷、硝酸锂、亚硝酸铷、亚硫酸钠、氰酸钠、氰酸锂、硫氰酸钠、硫氰酸钾、硬脂酸钠、硬脂酸铯、氢化钠、氢化钾、氢化锂、四苯硼钠、苯甲酸钠、苯甲酸锂、甲醇钠等碱金属的醇盐等,其中,从反应效率的观点出发,优选氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸铯等。另外,关于上述碱的添加量,只要使反应进行则没有特别限定,例如,相对于式(1-2)的金属络合物1摩尔,通常可以设定为0.05~10摩尔,从反应效率的观点出发,优选为0.08~6摩尔,更优选为0.10~5摩尔。反应中使用的溶剂没有特别限定,可以单独使用或组合使用例如戊烷、己烷、环戊烷、环己烷等脂肪族烃类、甲苯、二甲苯等芳香族烃类、二氯甲烷等卤代烃、丙酮、甲乙酮、环己酮等酮类溶剂、甲醇、乙醇、异丙醇、叔丁醇等醇类溶剂、四氢呋喃(THF)、乙醚、二甲氧基乙烷(DME)、二甲亚砜(DMSO)、二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、乙腈等有机溶剂。关于反应温度,只要使反应进行则没有特别限定,例如,通常可以设定为-40℃~20℃,从反应效率的观点出发,优选为-10℃~10℃,更优选为-5℃~5℃。关于反应时间,只要使反应进行则没有特别限定,例如,通常可以设定为0.1小时~30小时,从反应效率的观点出发,优选为0.1小时~2小时。上述迈克尔反应依赖于式(1-1)的金属络合物的手性轴的立体构型而高收率且高对映选择性地进行。即,在式(1-1)的金属络合物的手性轴的立体构型为S构型的情况下,通过迈克尔反应引入侧链时,α-氨基酸部分结构的α碳的立体构型成为R构型,在式(1-1)的金属络合物的手性轴的立体构型为R构型的情况下,α-氨基酸部分结构的α碳的立体构型成为S构型。(式中,R1、R2、R3、R4、R5、R6和M所示的符号与式(1-1)含义相同,斜体字的S或R表示立体构型,R16、R17、R18各自独立地表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基或卤素原子,R17和R18可以与它们所键合的碳原子一同形成环,EWG表示吸电子性基团)。作为R16、R17和R18所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基,可以列举例如R9中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。作为上述吸电子性基团,可以列举硝基(-NO2)、氰基(-CN)、甲苯磺酰基(-Ts)或甲磺酰基(-Ms)等有机磺酰基、氨磺酰基(-SO2NH2)、烷氧基羰基、芳氧基羰基、酰基、氨甲酰基(-CONH2)等羰基等。上述步骤[A-2]中的上述曼尼希反应是使式(1-1)的金属络合物与由伯胺或仲胺和醛生成的亚胺或亚铵根离子反应而在β碳上引入具有氨基的侧链的反应。本反应中,优选使用催化剂。本反应中使用的催化剂没有特别限定,可以使用本领域中通常使用的催化剂,可以列举例如二氮杂双环十一碳烯(DBU)、二氮杂双环壬烯(DBN)、三氮杂双环癸烯(TBD)、二氮杂双环[2.2.2]辛烷(DABCO)、L-脯氨酸或吡咯烷衍生物等。本反应通常使用溶剂来进行。本反应中使用的溶剂没有特别限定,可以使用本领域中通常使用的溶剂,可以使用四氢呋喃(THF)、二氧杂环己烷、二甲亚砜(DMSO)、乙腈等有机溶剂、水或者水与有机溶剂的任意比例的混合溶液中的任意一种。关于本反应中的反应温度,只要使反应进行则没有特别限定,例如,通常为-20℃~20℃,从反应效率的观点出发,优选为-10℃~10℃,更优选为-5℃~5℃。上述中说明的步骤[A-2]的反应均可高收率且高对映选择性地进行,也可以根据期望在步骤[A-2]的反应之后进行提高α-氨基酸部分结构的α碳的光学纯度的步骤。作为提高光学纯度的方法,没有特别限定,可以使用例如结晶化法(再结晶或悬浊等)、选择性溶解法、使用光学异构体分离用柱等的物理光学拆分等公知的方法。本发明的优选方式中,作为提高上述光学纯度的方法,采用如下方法:将上述步骤[A-2]中得到的金属络合物在甲醇、乙醇、异丙醇、叔丁醇、异丁醇等醇性溶剂中,在碳酸钾、碳酸钠、碳酸铯、氢氧化钾、氢氧化钠、氢氧化锂等碱的存在下,通常在40~80℃下加热0.5~24小时。通过加热,α-氨基酸部分结构的α碳的立体构型依赖于金属络合物的手性轴的立体构型而进行转换,因此化合物的光学纯度提高。接着,对整体线路图的步骤[A-3]的酸分解进行说明。通过对式(1-2)的金属络合物进行酸分解,可以使金属络合物中结合的α-氨基酸游离。作为上述酸分解中使用的酸,没有特别限定,只要是公知的酸即可,可以为无机酸,也可以为有机酸。作为上述无机酸,可以列举例如盐酸、硝酸、硫酸和高氯酸等,作为上述有机酸,可以列举例如乙酸、三氟乙酸、甲磺酸、三氟甲磺酸、草酸、丙酸、丁酸和戊酸等。其中,从能够高效地进行分解的观点出发,优选盐酸、硫酸、三氟乙酸或甲磺酸,更优选盐酸或甲磺酸。只要是可使酸分解充分进行的量,则上述酸的使用量没有特别限定,例如,相对于金属络合物1摩尔,通常可以设定为约0.1摩尔~约20摩尔,从分解效率的观点出发,优选设定为约0.3摩尔~约10摩尔。使用的溶剂优选醇类(甲醇、乙醇、异丙醇、叔丁醇等),优选使用甲醇、乙醇。溶剂的使用量没有特别限定,例如,相对于金属络合物1重量份,通常可以设定为约0.1倍容量~约100倍容量,优选为约0.5倍容量~约50倍容量。或者,溶剂的使用量相对于金属络合物1重量份,通常可以设定为约0.05重量份~约100重量份,优选为约0.1重量份~约50重量份。上述酸分解中的反应温度只要是能够在不引起化合物变性的情况下进行分解的温度,则没有特别限定,通常可以设定为约0℃~约100℃,从分解效率的观点出发,优选为约0℃~约80℃,更优选为约5℃~约60℃,特别优选为约40℃~约60℃。上述酸分解中的反应时间只要是充分进行分解的时间,则没有特别限定,通常可以设定为约0.1小时~约72小时,从分解效率的观点出发,优选为约0.1小时~约48小时,特别优选为约0.1小时~约20小时。关于压力,只要不阻碍反应则没有特别限定,例如可以设定为约0.1个大气压~约10个大气压。通过上述步骤[A-3],使下述式(6)所示的单取代的光学活性α-氨基酸或其盐游离。(式中,R9表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基或可以被取代的芳氧基羰基,**表示手性碳原子)。上述式(6)的手性碳原子(α碳)的立体构型依赖于上述步骤[A-2]中的侧链的引入时使用的式(1-1)的金属络合物的手性轴的立体构型。具体而言,关于通过上述烷基化反应得到的式(6)所示的α-氨基酸的立体构型,在式(1-1)的手性轴的立体构型为S构型的情况下成为D型,(式中,R9所示的符号与上述式(6)含义相同),在式(1-1)的手性轴的立体构型为R构型的情况下成为L型,(式中,R9所示的符号与上述式(6)含义相同)。另外,关于通过上述羟醛缩合反应得到的式(6)所示的光学活性α-氨基酸的α碳的立体构型,在式(1-1)的手性轴的立体构型为S构型的情况下成为S构型,(式中,R15所示的符号与上述含义相同),在式(1-1)的手性轴的立体构型为R构型的情况下成为R构型,(式中,R15所示的符号与上述含义相同)。此外,关于通过上述迈克尔反应得到的式(6)所示的光学活性α-氨基酸的α碳的立体构型,在式(1-1)的手性轴的立体构型为S构型的情况下成为R构型,(式中,R16、R17、R18和EWG所示的符号与上述含义相同),在式(1-1)的手性轴的立体构型为R构型的情况下成为S构型,(式中,R16、R17、R18和EWG所示的符号与上述含义相同)。在上述酸分解之后,可以利用公知的方法对游离出的光学活性α-氨基酸或其盐进行分离、纯化。关于公知的方法,只要是本领域中具有普通知识的人员可实施的方法则均可以采用,没有特别限定,可以采用例如使用离子交换树脂的方法。这样得到的光学活性α-氨基酸或其盐可以容易地转换为引入有适当保护基(例如,Z基、Fmoc基、Boc基等)的衍生物。在上述步骤[A-3]的酸分解之后,可以将式(3)的化合物回收、再利用。式(3)的回收率高(约90%以上)且不易发生光学纯度的降低,因此,能够高效地进行再利用。上述回收方法没有特别限定,可以采用本领域中通常使用的各种方法。可以采用例如使用转溶、浓缩、结晶化、层析的方法。本发明的一个方式中,可以在上述式(1-2)的金属络合物的α-氨基酸部分结构的α碳上再引入另一个侧链而制造式(2-1)的金属络合物。(式中,R1、R2、R3、R4、R5、R6、R9、M、*和**所示的符号与上述含义相同,R10表示可以被取代的烷基(例如,一部分或全部的氢原子被氟原子取代的烷基)、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基、可以被取代的烷氧基羰基或可以被取代的芳氧基羰基。或者,R9和R10可以与它们所键合的碳原子一同形成环)。作为R10所示的可以被取代的烷基、可以被取代的炔基、可以被取代的烯基、可以被取代的环烷基、可以被取代的芳基、可以被取代的杂芳基、可以被取代的芳烷基、可以被取代的杂芳基烷基,可以列举例如R9中所例示的基团等。作为此时的取代基的例子,可以列举关于R1所例示的取代基等。作为R9、R10与它们所键合的碳原子一同形成的环,没有特别限定,可以列举例如可以具有取代基的碳原子数3~12的环烷烃等。作为碳原子数3~12的环烷烃,可以列举例如环丙烷、环丁烷、环戊烷、环己烷、环己烯、十氢化萘、氧代环己烷、二氧代环己烷、羟基环戊烷、羟基环己烷等。作为此时的取代基的例子,可以列举R1中所例示的取代基等。上述在α碳上引入侧链的方法没有特别限定,可以采用例如烷基化反应、羟醛缩合反应、迈克尔反应、曼尼希反应等与亲电子剂的反应等。关于烷基化反应、羟醛缩合反应、迈克尔反应、曼尼希反应的各反应条件,除了将上述式(1-1)的金属络合物替换为式(1-2)以外,可以与上述[A-2]相同。该步骤相当于上述整体线路图的步骤[B-1]。需要说明的是,上述式(2-1)所示的金属络合物的一个优选方式为如下的金属络合物:R1为氢、氯、甲基或硝基,在两组R3和R4中的任意一组中,R3和R4均与所键合的芳香环的碳原子一同进一步形成芳香环或脂环式结构,R5和R6均为氢,R2为式(1-1a)所示的芳基。(R14表示氢原子或卤素原子)。通过对通过上述步骤[B-1]得到的式(2-1)的金属络合物进行酸分解,可以使金属络合物中结合的α-氨基酸游离。由此,能够得到具有所期望的手性的光学活性α,α-二取代α-氨基酸。关于酸分解的条件,除了将上述式(1-2)的化合物替换为式(2-1)以外,可以与上述步骤[A-3]相同。该步骤相当于上述整体线路图的步骤[B-2]。通过上述步骤[B-2],使下述式(7)所示的光学活性α,α-二取代α-氨基酸或其盐游离。(式中,R9、R10和**与上述式(2-1)含义相同)。所得到的上述(7)的光学活性α,α-二取代α-氨基酸或其盐的α碳与作为中间体的式(1-2)所示的金属络合物的手性轴的立体构型相应地优先地或选择性地具有S或R构型中的任意一种立体构型。(式中,R1、R2、R3、R4、R5、R6、R9、M、*和**所示的符号与上述含义相同)。需要说明的是,“优先地或选择性地”是指α碳的光学纯度达到约80%以上的高纯度。以下,在该说明书中是同样的。在上述酸分解之后,可以利用公知的方法对光学活性α,α-二取代α-氨基酸或其盐进行分离、纯化。关于公知的方法,只要是本领域中具有普通知识的人员可实施的方法则均可以采用。公知的方法没有特别限定,可以采用例如使用离子交换树脂的方法。这样得到的光学活性α,α-二取代α-氨基酸或其盐可以容易地转换为引入有适当保护基(例如,Z基、Fmoc基、Boc基等)的衍生物。在上述步骤[B-2]的酸分解之后,可以将式(3)的化合物回收、再利用。上述回收方法没有特别限定,可以采用本领域中通常使用的各种方法。可以采用例如使用转溶、浓缩、结晶化、层析的方法。本发明的另一个方式中,可以使用上述步骤[C-1]中得到的式(1-1’)所示的金属络合物而高收率且高对映选择性地制造式(8)所示的光学活性α-氨基酸。(式中,R1、R2、R3、R4、R5、R6、R11、*、M和**所示的符号与上述含义相同)。(式中,R10、R11和**与上述含义相同)。以下,对制造式(8)所示的光学活性α-氨基酸的方法进行具体说明。该步骤相当于上述的整体线路图的步骤[C-2]和[C-3]。该方式中,在上述式(1-1’)的金属络合物的α-氨基酸部分结构的α碳上引入侧链,得到式(2-1’)所示的金属络合物。(式中,R1~R6、R10、R11、M、*和**所示的符号与上述含义相同。或者,R10和R11可以与它们所键合的碳原子一同形成环)。式(2-1’)中,作为R10、R11与它们所键合的碳原子一同形成的环,可以列举式(2-1)中作为R9、R10与它们所键合的碳原子一同形成的环所例示的环。需要说明的是,上述式(2-1’)所示的金属络合物的一个优选方式为如下的金属络合物:R1为氢、氯、甲基或硝基,在两组R3和R4中的任意一组中,R3和R4均与所键合的芳香环的碳原子一同进一步形成芳香环或脂环式结构,R5和R6均为氢,R2为式(1-1a)所示的芳基。(R14表示氢原子或卤素原子)。上述在α碳上引入侧链的方法没有特别限定,可以采用例如烷基化反应、羟醛缩合反应、迈克尔反应、曼尼希反应等。关于烷基化反应、羟醛缩合反应、迈克尔反应、曼尼希反应的各反应条件,除了将上述式(1-1)的金属络合物替换为式(1-1’)以外,可以与上述[A-2]相同。该步骤相当于上述整体线路图的步骤[C-2]。通过对通过上述步骤[C-2]得到的式(2-1’)的金属络合物进行酸分解,可以使金属络合物中结合的α-氨基酸游离。由此,能够得到具有所期望的手性的上述式(8)的光学活性α-氨基酸或其盐。关于酸分解的条件,除了将上述式(1-2)的化合物替换为式(2-1’)以外,可以与上述步骤[A-3]相同。该步骤相当于上述整体线路图的步骤[C-3]。所得到的上述(8)的光学活性α-氨基酸或其盐的α碳的立体构型与式(1-1’)的金属络合物的手性轴的立体构型相应地优先地或选择性地具有S或R构型中的任意一种立体构型。在上述酸分解之后,可以利用公知的方法对式(8)的光学活性α-氨基酸或其盐进行分离、纯化。关于公知的方法,只要是本领域中具有普通知识的人员可实施的方法则均可以采用。公知的方法没有特别限定,可以采用例如使用离子交换树脂的方法。这样得到的式(8)的光学活性α-氨基酸或其盐可以容易地转换为引入有适当保护基(例如,Z基、Fmoc基、Boc基等)的衍生物。需要说明的是,在本说明书中,盐可以列举与例如盐酸、氢溴酸、硫酸、磷酸等无机酸、例如乙酸、三氟乙酸、甲磺酸、甲苯磺酸等有机酸、例如氢氧化钠、氢氧化钾等无机碱、例如三乙胺、环己胺等有机碱的盐。本发明的一个方式中,对于R9和R10与它们所键合的碳原子一同形成了环的式(2-1)所示的化合物、或者R10和R11与它们所键合的碳原子一同形成了环的式(2-1’)所示的化合物而言,可以使用例如式(1-2)所示的化合物或式(1-1’)所示的化合物作为中间体,通过例如烷基化反应、迈克尔反应等形成环;或者,也可以通过羟醛缩合反应等分子内缩合反应等形成环。作为环化反应,可以依照本领域中通常使用的公知方法、自身公知的方法或基于这些方法的方法来形成环。实施例接着,列举实验例、实施例进一步具体地对本发明进行说明,但本发明不受这些实施例的任何限定。实施例和参考例中,在以下的HPLC条件下进行测定。<HPLC条件-1:络合物分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=40:60~20:80(0~25分钟)20:80(25分钟~45分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:1.0mL/分钟温度:30℃检测器:UV254nm其他HPLC分析条件记载在各α-氨基酸及其衍生物的项目中。(实施例1)在部分结构中具有甘氨酸的Ni(II)络合物的合成实施例1-1:(S)-N-(2-苯甲酰基-4-氯苯基)-2-[3,5-二氢-4H-二萘并[2,1-c:1’,2’-e]氮杂-4-基]乙酰胺[手性助剂(S体)]在2-氨基-5-氯二苯甲酮(25.0g,107.9mmol)的乙腈溶液(500mL)中添加碳酸钾(44.7g,323.7mmol)和溴乙酰溴(28.3g,140.3mmol)的乙腈溶液(50mL),在室温下搅拌0.5小时。反应结束后,过滤沉淀,然后,将滤液浓缩干固。在浓缩残渣中加入自来水(75mL),利用乙酸乙酯(200mL,2次)分层。将有机层用自来水(150mL)洗涤,用硫酸钠干燥后,浓缩至150mL。在浓缩液中加入己烷(50mL),在室温下搅拌16小时后,在0℃下搅拌1小时,过滤析出的结晶。将结晶在30℃下真空干燥,以浅白色结晶的形式得到N-(2-苯甲酰基-4-氯苯基)-2-溴乙酰胺(33.16g,收率87%,化学纯度99.2%)。1H-NMR(200MHz,CDCl3):δ4.02(2H,s,COCH2),7.48-7.76(7H,m,ArH),8.55-8.60(1H,m,ArH),11.32(1H,brs,NH).在N-(2-苯甲酰基-4-氯苯基)-2-溴乙酰胺(2.0g,5.7mmol)的乙腈溶液(60mL)中添加碳酸钾(1.18g,8.5mmol)和(S)-联萘胺,加热至40℃,搅拌16小时。反应结束后,将反应悬浊液浓缩干固。将浓缩残渣利用硅胶层析(己烷:乙酸乙酯=4:1(v/v))进行纯化,以浅黄色结晶的形式得到(S)-N-(2-苯甲酰基-4-氯苯基)-2-[3,5-二氢-4H-二萘并[2,1-c:1’,2’-e]氮杂-4-基]乙酰胺(3.25g,收率为定量性的,化学纯度99.7%,99.8%ee)。ESI-MS(正离子模式):m/z=567.2,为[M+H]+.1H-NMR(200MHz,CDCl3):δ3.09和3.54(各1H,ABq,J=16.8Hz,COCH2),3.39和3.61(各2H,ABq,J=12.1Hz,2xNCH2),7.21-7.30(2H,m,ArH),7.42-7.65(11H,m,ArH),7.73-7.80(2H,m,ArH),7.92-7.98(2H,m,ArH),7.94(2H,d,J=8.2Hz,ArH),8.62(2H,d,J=8.6Hz,ArH),11.49(1H,brs,NH).13C-NMR(50.3MHz,CDCl3):δ56.4(CH2),60.3(CH2),123.3(ArCH),125.6(ArCH),125.9(ArCH),126.8(季ArC),127.5(ArCH),127.6(ArCH),127.8(季ArC),127.9(季ArC),128.3(ArCH),128.6(ArCH),128.7(ArCH),130.2(ArCH),131.4(季ArC),131.6(ArCH),133.1(ArCH),133.3(季ArC),135.0(季ArC),137.4(季ArC),137.6(季ArC),170.2(CO),196.4(CO).实施例1-2:在部分结构中具有甘氨酸的Ni(II)络合物[手性甘氨酸等价物(S体)]的合成在氩气气氛下,在(S)-N-(2-苯甲酰基-4-氯苯基)-2-[3,5-二氢-4H-二萘并[2,1-c:1’,2’-e]氮杂-4-基]乙酰胺(0.2g,0.353mmol)的甲醇溶液(30mL,甲醇在减压下超声波处理后吹入氩气40分钟以上而进行脱气处理)中添加乙酸镍·四水合物(0.176g,0.706mmol)、甘氨酸(0.132g,1.763mmol)、无水碳酸钾(0.439g,3.174mmol),回流1小时。反应结束后,在反应溶液中加入二氯甲烷(20mL)、自来水(20mL)、1N盐酸(5mL),分出有机层。将有机层用饱和盐水(12mL,3次)洗涤,用硫酸钠干燥后,浓缩干固,得到粗产物(0.259g)。使所得到的粗产物溶解在二氯甲烷(2mL)中,加入乙酸乙酯(2mL),静置使其结晶,将滤取的结晶在50℃下送风干燥,以红色结晶的形式得到在部分结构中具有甘氨酸的S型Ni(II)络合物(手性甘氨酸等价物(S体))(0.22g,收率91.7%)。ESI-MS(正离子模式):m/z=680.1,为[M+H]+.使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表1和图1。[表1]HPLC保留时间(分钟)19.54实施例1-3:在部分结构中具有甘氨酸的Ni(II)络合物[手性甘氨酸等价物(R体)]的合成在氩气气氛下,在(R)-N-(2-苯甲酰基-4-氯苯基)-2-[3,5-二氢-4H-二萘并[2,1-c:1’,2’-e]氮杂-4-基]乙酰胺(1.0g,1.763mmol)的甲醇溶液(150mL,甲醇在减压下超声波处理后吹入氩气40分钟以上而进行脱气处理)中添加乙酸镍·四水合物(0.878g,3.527mmol)、甘氨酸(0.662g,8.817mmol)、碳酸钾(2.194g,15.871mmol),回流1小时。反应结束后,将反应溶液稍微浓缩,加入二氯甲烷(100mL)、自来水(70mL)、1N盐酸(30mL),分出有机层。将有机层用自来水(60mL)、饱和盐水(60mL,2次)洗涤,用硫酸钠干燥后,浓缩干固,得到粗产物(1.182g)。使所得到的粗产物溶解在二氯甲烷(10mL)中,加入乙酸乙酯(10mL),静置使其结晶,将滤取的结晶在50℃下送风干燥,以红色结晶的形式得到在部分结构中具有甘氨酸的R型Ni(II)络合物(手性甘氨酸等价物(R体))(0.99g,收率82.4%)。ESI-MS(正离子模式):m/z=680.1,为[M+H]+.1H-NMR(200MHz,CDCl3):δ2.75[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.39[1H,d,J=15.9Hz,一个氮杂C(α’)H2N],3.65(2H,s,gly部分的CH2),3.75(1H,d,J=13.6Hz,一个乙酰苯胺NCOCH2),4.05[1H,d,J=15.9Hz,一个氮杂C(α’)H2N],4.07(1H,d,J=13.6Hz,一个乙酰苯胺NCOCH2),4.79[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.83(1H,d,J=2.4Hz),6.92-7.02(1H,m,ArH),7.03-7.13(1H,m,ArH),7.19-7.58(11H,m,ArH),7.92-8.02(3H,m,ArH),8.10(1H,d,J=8.3Hz,ArH),8.52(1H,d,J=9.2Hz,ArH),8.57(1H,d,J=8.3Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ58.7(NCOCH2),61.3(2xCH2),64.6(CH2),125.6(ArCH),125.7(ArCH),125.9(ArCH),126.1(ArCH),126.2(季ArC),126.37(ArCH),126.44(ArCH),126.8(季ArC),127.4(ArCH),127.6(ArCH),128.0(季ArC),128.4(ArCH),128.5(ArCH),128.9(ArCH),129.0(ArCH),129.8(ArCH),130.0(ArCH),130.1(ArCH),131.2(季ArC),132.2(ArCH),132.5(ArCH),133.7(季ArC),133.9(季ArC),135.7(季ArC),141.0(季ArC),171.4,174.8,176.6(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表2和图2。[表2]HPLC保留时间(分钟)19.68(实施例2)手性甘氨酸等价物的烷基化反应和光学活性α-氨基酸的合成实施例2-1:利用与溴化苄的烷基化反应进行的在部分结构中具有L-苯丙氨酸的Ni(II)络合物的合成在氩气气氛下在手性甘氨酸等价物(R体)(150.0mg,0.220mmol)的四氢呋喃(THF)溶液(2.6mL)中添加溶解在THF(0.4mL)中的溴化苄(41.5mg,0.242mmol)。然后,在氩气气氛下在0℃下滴加甲醇钠(35.7mg,0.661mmol)的甲醇溶液,在0℃下搅拌2小时。反应结束后,在反应液中加入水(10mL)、乙酸乙酯(10mL)进行分层,将水层用乙酸乙酯(10mL)萃取2次。将有机层用饱和盐水(10mL)洗涤,用硫酸钠干燥后,浓缩干固,得到红色固体(165.6mg)。在所得到的红色固体的甲醇溶液(3.3mL)中添加无水碳酸钾(59.4mg,0.644mmol),回流22小时。反应结束后,将反应液加入到冰冷却的0.5%乙酸水溶液(22mL)中,搅拌30分钟,使结晶析出,过滤,将滤取的结晶在50℃下送风干燥。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=40:1(v/v))进行纯化,以红色结晶的形式得到在部分结构中具有L-苯丙氨酸的Ni(II)络合物(130.9mg,收率77.1%,98.0%de)。ESI-MS(正离子模式):C46H35ClN3NiO3[M+H]+的m/z计算值770.17;实测值770.2.1H-NMR(200MHz,CDCl3):δ2.42[1H,d,J=12.1Hz,一个氮杂C(α)H2N],2.59(1H,ABX型的HA,JAB=13.6Hz,JAX=5.5Hz,一个Pheβ-CH2),2.61[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],2.76和3.17(各1H,ABq,J=13.9Hz,乙酰苯胺NCOCH2),3.00(1H,ABX型的HB,JAB=13.6Hz,JBX=3.0Hz,一个Pheβ-CH2),3.68[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.23(1H,ABX型的HX,JAX=5.5Hz,JBX=3.0Hz,Phe部分的α-H),4.54[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.67(1H,d,J=2.4Hz),7.05-8.02(21H,m,ArH),8.09(1H,d,J=8.4Hz,ArH),8.34(1H,d,J=9.2Hz,ArH),8.68(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ39.0(Phe部分的β-CH2),57.5(NCOCH2),61.6和65.9(氮杂的2xCH2),72.1(Phe部分的α-CH),125.2(ArCH),126.1(季ArC),126.4(ArCH),127.1(ArCH),127.4(ArCH),127.5(ArCH),127.7(ArCH),127.8(ArCH),128.4(ArCH),128.6(ArCH),128.8(季ArC),129.0(ArCH),129.1(ArCH),129.3(ArCH),129.4(ArCH),130.5(ArCH),131.0(季ArC),131.2(季ArC),131.4(季ArC),131.8(ArCH),132.4(ArCH),132.7(ArCH),132.9(季ArC),133.6(季ArC),133.9(季ArC),135.3(季ArC),135.9(季ArC),136.5(季ArC),141.4(季ArC),169.9,174.3,177.4(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表3和图3。[表3]实施例2-2:利用与4-氯溴苄的烷基化反应进行的在部分结构中具有D-4-氯苯丙氨酸的Ni(II)络合物的合成在手性甘氨酸等价物(S体)(100.0mg,0.147mmol)的四氢呋喃(THF)溶液(2.0mL)中添加4-氯溴苄(33.2mg,0.162mmol),接着在0℃下滴加甲醇钠(23.8mg,0.441mmol)的甲醇溶液,在0℃下搅拌5小时。反应结束后,在反应液中加入水(10mL)、乙酸乙酯(10mL),进行分层,将用乙酸乙酯(10mL)对水层萃取2次而得到的有机层用饱和盐水(10mL)洗涤,用硫酸钠干燥后,浓缩干固,得到橙红色固体(121.9mg)。在所得到的橙红色固体(116.3mg)的甲醇溶液(3.3mL)中添加无水碳酸钾(40.0mg,0.289mmol),回流24小时。反应结束后,在反应液中加入冰冷却的0.5%乙酸水溶液(21mL),搅拌30分钟,使结晶析出,将滤取的结晶在50℃下送风干燥。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=100:1(v/v))进行纯化,以红色结晶的形式得到在部分结构中具有D-4-氯苯丙氨酸的Ni(II)络合物(97.8mg,收率87%,99.8%de)。ESI-MS(正离子模式):C46H33Cl2N3NaNiO3[M+Na]+的m/z计算值826.12;实测值826.2.1H-NMR(200MHz,CDCl3):δ2.36[1H,d,J=12.3Hz,一个氮杂C(α)H2N],2.55(1H,ABX型的HA,JAB=13.6Hz,JAX=4.8Hz,一个4-Cl-Pheβ-CH2),2.69[1H,d,J=15.8Hz,一个氮杂C(α’)H2N],2.70和3.20(各1H,ABq,J=13.8Hz,乙酰苯胺NCOCH2),2.94(1H,ABX型的HB,JAB=13.6Hz,JBX=3.3Hz,一个p-Cl-Pheβ-CH2),3.74[1H,d,J=15.8Hz,一个氮杂C(α’)H2N],4.19(1H,ABX型的HX,JAX=4.8Hz,JBX=3.3Hz,p-Cl-Phe部分的α-H),4.59[1H,d,J=12.3Hz,一个氮杂C(α)H2N],6.58(1H,d,J=2.6Hz),6.95-7.07(2H,m,ArH),7.14-7.63(13H,m,ArH),7.64-7.72(2H,m,ArH),7.92-8.02(3H,m,ArH),8.11(1H,d,J=8.4Hz,ArH),8.39(1H,d,J=9.2Hz,ArH),8.83(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ38.5(p-Cl-Phe部分的β-CH2),57.9(NCOCH2),61.6和66.0(氮杂的2xCH2),71.8(p-Cl-Phe部分的α-CH),125.2(ArCH),126.1(季ArC),126.3(ArCH),126.4(ArCH),127.0(ArCH),127.3(ArCH),127.4(季ArC),127.8(ArCH),128.2(季ArC),128.5(ArCH),129.1(ArCH),129.2(ArCH),129.4(ArCH),130.5(ArCH),130.9(季ArC),131.1(季ArC),131.4(季ArC),132.4(ArCH),132.8(ArCH),133.0(ArCH),133.6(季ArC),133.86(季ArC),133.93(季ArC),134.8(季ArC),135.1(季ArC),136.0(季ArC),141.4(季ArC),170.2,174.4,177.0(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表4和图4。[表4]实施例2-3:在部分结构中具有D-4-氯苯丙氨酸的Ni(II)络合物在酸条件下的D-4-氯苯丙氨酸的游离和利用Boc基的保护向在部分结构中具有D-4-氯苯丙氨酸的Ni(II)络合物(92.5mg,0.115mmol)的甲醇(2.8mL)悬浊液中添加1N盐酸(0.6mL,0.574mmol),在40℃下搅拌6小时。反应结束后,将反应液浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL)进行分层。分取水层,蒸馏除去溶剂后,将所得到的固体溶解到9%氨水(3mL)中,通入到阳离子交换树脂柱[SK-1B,18mL,洗脱液:2~4%氨水]中,得到D-4-氯苯丙氨酸粗产物(22.0mg,收率96%)。另一方面,将有机层用4%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(S体)(62.5mg,收率96%)。将D-4-氯苯丙氨酸粗产物(22.0mg,0.110mmol)溶解到水(2mL)和丙酮(1.0mL)中,添加(Boc)2O(40.4mg,0.185mmol)的丙酮(0.5mL)溶液和三乙胺(18.8mg,0.186mmol)的丙酮(0.5mL)溶液,在室温下搅拌44小时。将反应液浓缩至2mL以下后,加入甲苯(5mL),在搅拌下加入4N盐酸直至水层的pH为2~3。分取有机层,用饱和盐水(5mL,2次)洗涤,用硫酸镁干燥后,蒸馏除去溶剂,以白色固体的形式得到Boc-D-4-氯苯丙氨酸(28.7mg,87.3%,98.6%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表5和图5。<HPLC条件:Boc-D-4-Cl-Phe手性分析条件>柱:CHIRALPAKAD-RH(5μm,150×4.6mmi.d.)洗脱液:A:B=35:65A=0.1%磷酸水溶液B=0.1%磷酸乙腈溶液流量:1.0mL/分钟温度:35℃检测器:UV254nm[表5]实施例2-4:利用与2-(溴甲基)萘的烷基化反应进行的在部分结构中具有3-(2-萘基)-D-丙氨酸的Ni(II)络合物的合成在氩气气氛下,在0℃下在手性甘氨酸等价物(S体)(200.0mg,0.294mmol)的四氢呋喃(THF)溶液(4.0mL)中添加2-(溴甲基)萘(71.5mg,0.323mmol),接着滴加甲醇钠(95.2mg,1.763mmol)的甲醇溶液,在0℃下搅拌1.5小时。在反应液中加入水(10mL)和乙酸乙酯(10mL)进行分层,将水层用乙酸乙酯(10mL)萃取3次。将全部有机层用饱和盐水(10mL)洗涤,用硫酸钠干燥后,浓缩干固,得到橙红色固体(275.9mg)。在所得到的橙红色固体(266.6mg)的甲醇溶液(5.4mL)中添加无水碳酸钾(89.8mg,0.650mmol),回流24小时。将反应液加入到冰冷却的0.5%乙酸水溶液(50mL)中,搅拌30分钟,滤取析出的结晶,在50℃下送风干燥。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=50:1(v/v))进行纯化,以红色结晶的形式得到在部分结构中具有3-(2-萘基)-D-丙氨酸的Ni(II)络合物(232.9mg,87.4%,化学纯度97.9%,99.8%de)。ESI-MS(正离子模式):C50H37ClN3NiO3[M+H]+的m/z计算值820.19;实测值820.31H-NMR(200MHz,CDCl3):δ1.84(1H,d,J=13.9Hz,一个乙酰苯胺NCOCH2),2.12[1H,d,J=12.3Hz,一个氮杂C(α)H2N],2.13[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],2.54[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],2.74(1H,ABX型的HA,JAB=13.6Hz,JAX=5.0Hz,一个AAβ-CH2),2.92(1H,d,J=13.9Hz,一个乙酰苯胺NCOCH2),3.15(1H,ABX型的HB,JAB=13.6Hz,JBX=2.9Hz,一个AAβ-CH2),4.25(1H,ABX型的HX,JAX=5.0Hz,JBX=2.9Hz,AA部分的α-H),4.43[1H,d,J=12.3Hz,一个氮杂C(α)H2N],6.52(1H,d,J=8.4Hz,ArH),6.61(1H,d,J=2.6Hz,ArH),7.01(1H,brd,J=7.7Hz,ArH),7.09-7.33(6H,m,ArH),7.38-8.14(15H,m,ArH),8.21(1H,brd,J=7.9Hz,ArH),8.34(1H,d,J=9.2Hz,ArH),8.75(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ39.1(Phe部分的β-CH2),57.1(NCOCH2),61.5和65.3(氮杂的2xCH2),72.3(AA部分的α-CH),125.2(ArCH),126.0(季ArC),126.2(ArCH),126.3(ArCH),126.7(ArCH),126.9(ArCH),127.1(ArCH),127.3(ArCH),127.4(ArCH),127.5(ArCH),127.8(ArCH),128.0(ArCH),128.2(季ArC),128.3(ArCH),128.4(ArCH),128.5(ArCH),128.7(ArCH),129.1(ArCH),129.3(ArCH),129.4(ArCH),130.0(ArCH),130.4(ArCH),130.9(季ArC),131.0(季ArC),131.4(季ArC),132.3(ArCH),132.6(ArCH),132.9(季ArC),133.2(季ArC),133.4(季ArC),133.9(季ArC),134.0(季ArC),135.0(季ArC),135.9(季ArC),141.5(季ArC),169.9,174.3,177.3(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表6和图6。[表6]实施例2-5:在部分结构中具有3-(2-萘基)-D-丙氨酸的Ni(II)络合物在酸条件下的3-(2-萘基)-D-丙氨酸的游离和利用Boc基的保护向在部分结构中具有3-(2-萘基)-D-丙氨酸的Ni(II)络合物(100.0mg,0.122mmol)的甲醇(4.0mL)悬浊液中添加1N盐酸(0.61mL,0.609mmol),在40-50℃下搅拌8小时。将反应液浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL)进行分层。分取水层,蒸馏除去溶剂后,将所得到的固体溶解到9%氨水(3mL)中,通入到阳离子交换树脂柱[SK-1B,21mL,洗脱液:2~4%氨水]中,以白色固体的形式得到3-(2-萘基)-D-丙氨酸粗产物(22.0mg,收率83.9%)。另一方面,将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(S体)(58.7mg,化学纯度98.5%,收率85.0%)。将3-(2-萘基)-D-丙氨酸粗产物(13.0mg,0.060mmol)溶解到水(2mL)和丙酮(1.0mL)中,添加(Boc)2O(21.1mg,0.097mmol)的丙酮(0.5mL)溶液和三乙胺(9.8mg,0.097mmol)的丙酮(0.5mL)溶液,在室温下搅拌27小时。将反应液浓缩至2mL以下后,加入甲苯(5mL),在搅拌下加入1N盐酸直至水层的pH为2~3。将水层用甲苯(5mL,3次)萃取,将所得到的有机层用饱和盐水(5mL,2次)洗涤,用硫酸镁干燥后,蒸馏除去溶剂,以无色固体的形式得到Boc-3-(2-萘基)-D-丙氨酸(16.2mg,收率85.1%,化学纯度96.6%,99.3%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表7和图7。<HPLC条件:Boc-3-(2-萘基)-D-丙氨酸手性分析条件>柱:CHIRALPAKAD-RH(5μm,150×4.6mmi.d.)洗脱液:A:B=35:65A=0.1%磷酸水溶液B=0.1%磷酸乙腈溶液流量:0.5mL/分钟温度:30℃检测器:UV220nm[表7]实施例2-6:利用与3-(溴甲基)吡啶的烷基化反应进行的在部分结构中具有3-(3-吡啶基)-D-丙氨酸的Ni(II)络合物的合成在氩气气氛下,在0℃下在手性甘氨酸等价物(S体)(200.0mg,0.294mmol)的四氢呋喃(THF)溶液(4.0mL)中添加3-(溴甲基)吡啶氢溴酸盐(81.8mg,0.323mmol),接着滴加甲醇钠(238.0mg,4.41mmol)的甲醇溶液,在0℃下搅拌1小时。在反应液中加入水(15mL)和乙酸乙酯(15mL)进行分层,将水层用乙酸乙酯(10mL)萃取3次。将有机层用饱和盐水(10mL)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,得到橙红色固体(238.0mg)。在该橙红色固体(231.1mg)的甲醇溶液(2.0mL)中添加无水碳酸钾(82.8mg,0.599mmol),在氩气气氛下在40℃下搅拌24小时。将反应液加入到冰冷却的0.5%乙酸水溶液(20mL)中,搅拌30分钟,滤取析出的结晶后,在50℃下送风干燥。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=9:1(v/v))进行纯化,以红色结晶的形式得到在部分结构中具有3-(3-吡啶基)-D-丙氨酸的Ni(II)络合物(171.2mg,77.8%,化学纯度97.9%,99.3%de)。ESI-MS(正离子模式)C45H34ClN4NiO3[M+H]+的m/z计算值771.17;实测值771.21H-NMR(200MHz,CDCl3):δ2.42[1H,d,J=12.1Hz,一个氮杂C(α)H2N],2.61(1H,ABX型的HA,JAB=13.8Hz,JAX=5.3Hz,一个AAβ-CH2),2.69[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],2.87(1H,d,J=13.8Hz,一个乙酰苯胺NCOCH2),3.02(1H,ABX型的HB,JAB=13.8Hz,JBX=2.6Hz,一个AAβ-CH2),3.27(1H,d,J=13.8Hz,一个乙酰苯胺NCOCH2),3.80[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.25(1H,ABX型的HX,JAX=5.3Hz,JBX=2.6Hz,AA部分的α-H),4.56[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.64(1H,d,J=2.4Hz),7.02-7.65(15H,m,ArH),7.92-8.01(3H,m,ArH),8.11(1H,d,J=8.4Hz,ArH),8.39(1H,d,J=9.2Hz,ArH),8.74(1H,d,J=8.2Hz,ArH),8.95(1H,brs,ArH),9.03(1H,brd,J=2.7Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ36.4(β-CH2),57.8(NCOCH2),61.6和65.8(氮杂的2xCH2),71.3(α-CH),124.0(ArCH),125.2(ArCH),126.1(季ArC),126.3(ArCH),126.4(ArCH),127.1(ArCH),127.3(ArCH),127.4(ArCH),127.7(ArCH),128.0(季ArC),128.4(ArCH),128.6(ArCH),129.0(ArCH),129.2(ArCH),129.5(ArCH),130.6(ArCH),130.8(季ArC),131.1(季ArC),131.4(季ArC),132.1(季ArC),132.4(ArCH),132.8(季ArC),132.9(ArCH),133.6(季ArC),133.9(季ArC),135.2(季ArC),136.0(季ArC),138.7(季ArC),141.5(季ArC),149.2(季ArC),152.2(季ArC),170.5,174.3,177.0(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表8和图8。[表8]实施例2-7:在部分结构中具有3-(3-吡啶基)-D-丙氨酸的Ni(II)络合物在酸条件下的3-(3-吡啶基)-D-丙氨酸的游离和利用Boc基的保护向在部分结构中具有3-(3-吡啶基)-D-丙氨酸的Ni(II)络合物(100.0mg,0.130mmol)的甲醇(3.0mL)悬浊液中添加1N盐酸(0.65mL,0.648mmol),在室温下搅拌3小时。将反应液在减压下浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL)进行分层。分取水层,蒸馏除去溶剂后,将所得到的固体溶解到8%氨水(5mL)中,通入到阳离子交换树脂柱(SK-1B,40mL,洗脱液:水、接着为4%氨水),以白色固体的形式得到3-(3-吡啶基)-D-丙氨酸(17.7mg,收率81.9%,97.3%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表9和图9。<HPLC条件:3-(3-吡啶基)-D-丙氨酸手性分析条件>柱:CROWNPAKCR(+)(5μm,150×4.0mmi.d.)洗脱液:高氯酸水溶液(pH1.0)流量:0.4mL/分钟温度:30℃检测器:UV254nm[表9]另一方面,将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(S体)(63.8mg,化学纯度98.6%,收率86.9%)。将3-(3-吡啶基)-D-丙氨酸(16.3mg,0.098mmol)溶解到水(1mL)和丙酮(0.5mL)中,添加(Boc)2O(34.3mg,0.157mmol)的丙酮(0.25mL)溶液和三乙胺(15.9mg,0.157mmol)的丙酮(0.25mL)溶液,在室温下搅拌3.5小时。将反应液在减压下浓缩至1mL以下后,加入2-丁醇(10mL),在搅拌下加入1N盐酸直至水层的pH为2~3。将水层用2-丁醇(10mL,3次)萃取,将所得到的有机层用饱和盐水(10mL,2次)洗涤,用硫酸镁干燥后,蒸馏除去溶剂,以无色固体的形式得到Boc-3-(3-吡啶基)-D-丙氨酸(20.5mg,收率78.5%,化学纯度97.1%)。实施例2-8:利用与烯丙基溴的烷基化反应进行的在部分结构中具有L-烯丙基甘氨酸的Ni(II)络合物的合成在氩气气氛下,在手性甘氨酸等价物(R体)(171.9mg,0.253mmol)的四氢呋喃(THF)溶液(3.5mL)中,在0℃下添加烯丙基溴(33.6mg,0.278mmol),接着滴加甲醇钠(40.9mg,0.758mmol)的甲醇溶液,在0℃下搅拌1小时。在反应液中加入水(10mL)和乙酸乙酯(10mL)进行分层,将水层用乙酸乙酯(10mL)萃取2次。将全部有机层用饱和盐水(10mL)洗涤,用硫酸钠干燥后,浓缩干固,得到橙红色固体(184.5mg)。在所得到的橙红色固体(184.5mg)的甲醇溶液(3.7mL)中添加无水碳酸钾(70.8mg,0.512mmol),在氩气气氛下在40℃下搅拌3.5小时。将反应液加入到冰冷却的0.5%乙酸水溶液(37mL)中,搅拌30分钟。滤取析出的结晶,在50℃下送风干燥。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=50:1(v/v))进行纯化,以红色结晶的形式得到在部分结构中具有L-烯丙基甘氨酸的Ni(II)络合物(144.9mg,收率78.6%,化学纯度98.2%,98.6%de)。ESI-MS(正离子模式):C42H33ClN3NiO3[M+H]+的m/z计算值720.16;实测值720.21H-NMR(200MHz,CDCl3):δ2.23(1H,ddd,J=13.9,7.9,5.9Hz,烯丙基-Gly部分的一个β-CH2),2.39-2.53(1H,m,烯丙基-Gly部分的一个β-CH2),2.65[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.02[1H,d,J=15.4Hz,一个氮杂C(α’)H2N],3.60和3.69(各1H,ABq,J=13.9Hz,乙酰苯胺NCOCH2),3.94(1H,dd,J=5.9,3.7Hz,烯丙基-Gly部分的α-H),4.49[1H,d,J=15.4Hz,一个氮杂C(α’)H2N],4.76[1H,d,J=12.1Hz,一个氮杂C(α)H2N],5.35(1H,dd,J=17.0,1.5Hz),5.74(1H,dd,J=10.1,1.5Hz),6.60-6.95(2H,m),6.63(1H,d,J=2.6Hz,ArH),7.09-7.15(1H,m,ArH),7.20-7.58(11H,m,ArH),7.92-8.02(3H,m,ArH),8.14(1H,d,J=8.2Hz,ArH),8.40(1H,d,J=9.0Hz,ArH),8.78(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ38.2(CH2),59.2(CH2),61.6和66.6(氮杂的2xCH2),70.8(α-CH),120.4(=CH2),125.1(ArCH),126.2(季ArC),126.3(季ArC),126.4(ArCH),127.0(ArCH),127.3(ArCH),127.4(ArCH),127.5(ArCH),127.9(ArCH),128.4(ArCH),128.5(季ArC),128.7(ArCH),129.1(ArCH),129.3(ArCH),129.5(ArCH),130.3(ArCH),131.0(季ArC),131.2(季ArC),131.5(季ArC),132.4(ArCH),132.6(ArCH),132.8(季ArC),133.7(季ArC),134.0(季ArC),135.5(季ArC),136.0(季ArC),141.1(季ArC),170.0,174.3,177.6(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表10和图10。<HPLC条件-1:络合物分析条件>[表10]实施例2-9:在部分结构中具有L-烯丙基甘氨酸的Ni(II)络合物在酸条件下的L-烯丙基甘氨酸的游离和利用Boc基的保护在氩气气氛下,向在部分结构中具有L-烯丙基甘氨酸的Ni(II)络合物(107.6mg,0.149mmol)的甲醇(3.3mL)悬浊液中添加1N盐酸(0.75mL,0.75mmol),在40℃下搅拌2小时。反应结束后,将反应液浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL)进行分层。分取水层,蒸馏除去溶剂后,将所得到的固体溶解到4%氨水(3mL)中,通入到阳离子交换树脂柱(SK-1B,18mL,洗脱液:水、接着为2-4%氨水)中,定量地得到L-烯丙基甘氨酸(18.9mg,96.9%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表11和图11。<HPLC条件:L-烯丙基甘氨酸手性分析条件>柱:CROWNPAKCR(+)(5μm,150×4.0mmi.d.)洗脱液:高氯酸水溶液(pH2.0)流量:0.5mL/分钟温度:20℃检测器:UV200nm[表11]另一方面,将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,定量地回收手性助剂(R体)(94.2mg,化学纯度98.1%)。将L-烯丙基甘氨酸(17.7mg,0.154mmol)溶解到水(2mL)和丙酮(1.0mL)中,添加(Boc)2O(36.9mg,0.169mmol)的丙酮(0.5mL)溶液和三乙胺(17.1mg,0.169mmol)的丙酮(0.5mL)溶液,在室温下搅拌22小时。将反应液浓缩至2mL以下后,加入甲苯(5mL),在搅拌下加入1N盐酸直至水层的pH为2~3。将水层用甲苯(5mL,3次)萃取,将全部有机层用饱和盐水(5mL,2次)洗涤,用硫酸镁干燥后,蒸馏除去溶剂。将浓缩残渣利用硅胶柱层析(二氯甲烷-甲醇)进行纯化,以无色油状物质的形式得到Boc-L-烯丙基甘氨酸(18.7mg,56.5%,化学纯度95.5%)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表12。<HPLC条件:Boc-L-烯丙基甘氨酸分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=80:20~20:80(0~25分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:1.0mL/分钟温度:30℃检测器:UV200nm[表12](实施例3)光学活性的α,α-二取代α-氨基酸的合成参考例1:在部分结构中具有丙氨酸的Ni(II)络合物的合成在手性助剂(R体)(0.2g,0.353mmol)的甲醇悬浊液(4mL)中添加乙酸镍·四水合物(0.176g,0.706mmol)、DL-丙氨酸(0.063g,0.706mmol)、碳酸钾(0.293g,2.118mmol),在40℃下加热24小时。反应结束后,将反应液加入到冰冷却的5%乙酸水溶液(30mL)中,搅拌30分钟,使结晶析出,过滤。将滤取的结晶在50℃下送风干燥,以红色结晶的形式得到在部分结构中具有丙氨酸的Ni(II)络合物(0.207g,收率84.8%,96%de)。ESI-MS(正离子模式):m/z=694.2,为[M+H]+.1H-NMR(200MHz,CDCl3):δ1.51(3H,d,J=7.0Hz,Me),2.73[1H,d,J=12.2Hz,一个氮杂C(α)H2N],3.08[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.68和3.76(各1H,ABq,J=13.9Hz,乙酰苯胺NCOCH2),3.84(1H,q,J=7.0Hz,Ala部分的α-H),4.57[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.84[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.66(1H,d,J=2.6Hz),6.91-6.99(1H,m,ArH),7.16-7.32(4H,m,ArH),7.35-7.41(1H,m,ArH),7.43-7.57(7H,m,ArH),7.94-8.03(3H,m,ArH),8.16(1H,d,J=8.3Hz,ArH),8.44(1H,d,J=9.2Hz,ArH),8.76(1H,d,J=8.3Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ21.5(Ala部分的Me),58.7(NCOCH2),61.9和66.3(氮杂的2xCH2),66.9(Ala部分的α-CH),125.1(ArCH),126.1(季ArC),126.37(季ArC),126.44(ArCH),126.9(ArCH),127.3(ArCH),127.4(ArCH),127.5(ArCH),127.6(ArCH),127.8(ArCH),128.2(季ArC),128.4(ArCH),128.7(ArCH),129.2(ArCH),129.5(ArCH),130.2(ArCH),131.0(季ArC),131.3(季ArC),131.5(季ArC),132.4(ArCH),132.6(ArCH),132.7(季ArC),133.7(季ArC),134.1(季ArC),135.6(季ArC),136.0(季ArC),140.9(季ArC),170.2,174.6,179.7(CN和2xCO).实施例3-1:利用在部分结构中具有丙氨酸的Ni(II)络合物与溴化苄的反应进行的在部分结构中具有α-甲基-L-苯丙氨酸的Ni(II)络合物的合成在氩气气氛下,向在部分结构中具有丙氨酸的Ni(II)络合物(258.6mg,0.372mmol)的四氢呋喃(THF)溶液(5.2mL)中依次加入溴化苄(70.0mg,0.409mmol)和氢化钠(26.8mg,1.117mmol),在室温(23-24℃)下搅拌2.0小时。在反应液中加入水(10mL)和乙酸乙酯(10mL),搅拌,分层后,将水层用乙酸乙酯(10mL)萃取3次。将全部有机层用饱和盐水(15mL)洗涤,用硫酸钠(10g)干燥后,蒸馏除去溶剂,得到橙红色固体(302.8mg)。将所得到的橙红色固体(302.8mg)在二氯甲烷:乙酸乙酯=1:1(10v/w)中进行再结晶,以红色结晶的形式得到在部分结构中具有α-甲基-L-苯丙氨酸的Ni(II)络合物(215.6mg,收率73.8%,化学纯度99.4%,>99.9%de)。ESI-MS(正离子模式):C47H37ClN3NiO3[M+H]+的m/z计算值784.19;实测值784.21H-NMR(200MHz,CDCl3):δ1.01(3H,s,α-Me),2.46[1H,d,J=12.1Hz,一个氮杂C(α)H2N],2.57[1H,d,J=15.5Hz,一个氮杂C(α’)H2N],2.71(1H,d,J=13.9Hz,一个α-MePheβ-CH2),2.97和3.03(各1H,ABq,J=13.6Hz,乙酰苯胺NCOCH2),3.27(1H,d,J=13.9Hz,一个α-MePheβ-CH2),3.52[1H,d,J=15.5Hz,一个氮杂C(α’)H2N],4.55[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.73(1H,d,J=2.4Hz),7.12-7.36(7H,m,ArH),7.40-7.62(8H,m,ArH),7.67-7.86(3H,m,ArH),7.87-7.98(3H,m,ArH),8.07(1H,d,J=8.2Hz,ArH),8.23(1H,d,J=9.2Hz,ArH),8.63(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ29.2(Me),47.4(β-CH2),57.6(NCOCH2),61.7和65.6(氮杂的2xCH2),80.4(季),124.8(ArCH),125.8(季ArC),126.3(ArCH),126.5(ArCH),127.2(ArCH),127.4(ArCH),127.6(ArCH),127.9(ArCH),128.37(ArCH),128.44(ArCH),128.7(ArCH),128.9(ArCH),129.0(ArCH),129.3(ArCH),129.9(ArCH),131.1(季ArC),130.3(ArCH),131.1(季ArC),131.4(季ArC),131.7(ArCH),132.3(ArCH),132.9(ArCH),133.5(季ArC),133.8(季ArC),135.3(季ArC),135.8(季ArC),136.0(季ArC),137.6(季ArC),140.6(季ArC),170.9,174.1,179.4(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表13和图12。[表13]实施例3-2:在部分结构中具有α-甲基-L-苯丙氨酸的Ni(II)络合物在酸条件下的α-甲基-L-苯丙氨酸的游离和利用Boc基的保护向在部分结构中具有α-甲基-L-苯丙氨酸的Ni(II)络合物(190.0mg,0.242mmol)的甲醇(5.7mL)悬浊液中添加1N盐酸(1.2mL,1.20mmol,5eq.),在40-50℃下搅拌5小时。将反应液在减压下浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL),将其溶解并分层后,将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,定量地回收手性助剂(R体)(138.2mg,化学纯度92.2%)。另一方面,将水层浓缩干固,将所得到的固体溶解到8%氨水(0.5mL)和甲醇(5.0mL)的混合溶剂中,通入到阳离子交换树脂柱(SK-1B,40mL,洗脱液:水、接着为4%氨水)中,以白色固体的形式得到α-甲基-L-苯丙氨酸(25.6mg,收率59.0%)。将所得到的α-甲基-L-苯丙氨酸(20.0mg,0.112mmol)悬浊到脱水乙腈(1.0mL)中,加入四甲基氢氧化铵五水合物(20.2mg,0.112mmol),在室温下搅拌1小时,加入(Boc)2O(36.5mg,0.167mmol),在室温下搅拌76小时。将反应液浓缩,将所得到的残渣溶解到水(5mL)中,用异丙醚(2mL)洗涤2次后,在水层中添加柠檬酸,使pH为3后,将水层用乙酸乙酯(10mL)萃取3次。将全部有机层用水(5mL,2次)和饱和盐水(5mL,2次)洗涤,用硫酸镁干燥后,蒸馏除去溶剂,以无色油状物质的形式得到Boc-α-甲基-L-苯丙氨酸(29.3mg,55.5%,化学纯度91.3%,99.6%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表14和图13。<HPLC条件:Boc-α-甲基-L-苯丙氨酸手性分析条件>柱:CHIRALPAKAY-H(5μm,150×4.6mmi.d.)洗脱液:己烷/乙醇=92.5/7.5,0.3%三氟乙酸流量:1.0mL/分钟温度:40℃检测器:UV220nm[表14]实施例3-3:利用在部分结构中具有丙氨酸的Ni(II)络合物与4-氟溴苄的反应进行的在部分结构中具有4-氟-α-甲基-L-苯丙氨酸的Ni(II)络合物的合成在氩气气氛下,向在部分结构中具有丙氨酸的Ni(II)络合物(200.0mg,0.288mmol)的四氢呋喃(THF)溶液(4mL)中依次加入4-氟溴苄(59.9mg,0.317mmol)和氢化钠(20.8mg,0.864mmol),在室温(23℃)下搅拌2小时。在反应液中加入水(10mL)和乙酸乙酯(10mL),搅拌,分层后,将水层用乙酸乙酯(10mL)萃取3次。将全部有机层用饱和盐水洗涤,用硫酸钠干燥后,蒸馏除去溶剂,将所得到的橙红色固体(225.0mg)利用硅胶柱层析(二氯甲烷:丙酮=50:1(v/v))进行纯化,接着从二氯甲烷:甲醇=1:5(16v/w)中再结晶,以红色结晶的形式得到在部分结构中具有4-氟-α-甲基-L-苯丙氨酸的Ni(II)络合物(107.4mg,收率46.5%,化学纯度99.7%,>99.9%de)。ESI-MS(正离子模式):C47H36ClFN3NiO3[M+H]+的m/z计算值802.18;实测值802.31H-NMR(200MHz,CDCl3):δ1.02(3H,s,α-Me),2.54[1H,d,J=12.1Hz,一个氮杂C(α)H2N],2.690[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],2.694(1H,d,J=13.7Hz,一个α-Me-4-氟Pheβ-CH2),2.96和2.99(各1H,ABq,J=13.8Hz,乙酰苯胺NCOCH2),3.32(1H,d,J=13.7Hz,一个α-Me-4-氟Pheβ-CH2),3.59[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.62[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.76(1H,d,J=2.6Hz,ArH),7.09-7.15(1H,m,ArH),7.18-7.59(16H,m,ArH),7.88-8.01(3H,m,ArH),8.06(1H,d,J=8.2Hz,ArH),8.26(1H,d,J=9.2Hz,ArH),8.57(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ29.2(Me),46.5(β-CH2),58.1(NCOCH2),61.9和65.7(氮杂的2xCH2),80.3(季),115.8(d,3JCF=22.0Hz),124.9(ArCH),126.0(季ArC),126.3(ArCH),126.4(ArCH),126.5(ArCH),127.3(ArCH),127.5(ArCH),127.7(ArCH),127.8(ArCH),128.4(ArCH),128.7(季ArC),128.8(ArCH),129.2(ArCH),129.4(ArCH),130.0(季ArC),130.2(d,4JCF=14.6Hz),131.1(季ArC),131.2(季ArC),131.4(季ArC),132.4(ArCH),132.9(ArCH),133.1(ArCH),133.2(ArCH),133.3(季ArC),133.6(季ArC),133.9(季ArC),135.2(季ArC),135.8(季ArC),136.0(季ArC),140.6(季ArC),162.9(d,1JCF=247.1Hz),171.2,174.1,179.3(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表15和图14。[表15]实施例3-4:利用在部分结构中具有丙氨酸的Ni(II)络合物与烯丙基溴的反应进行的在部分结构中具有(S)-α-烯丙基丙氨酸的Ni(II)络合物的合成在氩气气氛下,向在部分结构中具有丙氨酸的Ni(II)络合物(500.0mg,0.720mmol)的四氢呋喃(THF)溶液(10mL)中依次加入烯丙基溴(95.8mg,0.792mmol)和氢化钠(51.8mg,2.129mmol),在室温(23℃)下搅拌1小时。在反应液中加入水(20mL)和乙酸乙酯(20mL)进行分层,将水层用乙酸乙酯(10mL)萃取3次。将全部有机层用饱和盐水洗涤,用硫酸钠(7g)干燥后,浓缩干固,将所得到的粗产物(528.5mg)从二氯甲烷:乙酸乙酯=1:1.5中进行再结晶,以红色结晶的形式得到在部分结构中具有(S)-α-烯丙基丙氨酸的Ni(II)络合物(346.3mg,收率65.5%,化学纯度96.8%,98.7%de)。ESI-MS(正离子模式):C43H35ClN3NiO3[M+H]+的m/z计算值734.17;实测值734.01H-NMR(200MHz,CDCl3):δ1.01(3H,s,α-Me),2.45(1H,ABX型的HA,JAB=13.9Hz,JAX=7.3Hz,烯丙基-Ala部分的一个β-CH2),2.53(1H,ABX型的HB,JAB=13.9Hz,JBX=7.1Hz,烯丙基-Ala部分的一个β-CH2),2.76[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.12[1H,d,J=15.4Hz,一个氮杂C(α’)H2N],3.55和3.80(各1H,ABq,J=13.9Hz,乙酰苯胺NCOCH2),4.38[1H,d,J=15.4Hz,一个氮杂C(α’)H2N],4.79[1H,d,J=12.1Hz,一个氮杂C(α)H2N],5.49(1H,dd,J=17.0,1.5Hz),5.79(1H,dd,J=10.1,1.5Hz),6.75(1H,d,J=2.4Hz,ArH),6.88-7.10(2H,m),7.20-7.57(12H,m,ArH),7.92-8.03(3H,m,ArH),8.11(1H,d,J=8.2Hz,ArH),8.30(1H,d,J=9.0Hz,ArH),8.67(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ28.6(Me),45.3(CH2),59.0(CH2),61.5和66.0(氮杂的2xCH2),78.9(季),120.1(=CH2),124.8(ArCH),126.0(季ArC),126.2(ArCH),126.4(ArCH),126.8(ArCH),127.46(ArCH),127.53(ArCH),128.0(ArCH),128.4(ArCH),128.5(ArCH),128.7(季ArC),129.1(ArCH),129.4(ArCH),129.8(ArCH),129.9(ArCH),130.4(季ArC),131.17(季ArC),131.24(季ArC),131.4(季ArC),132.2(ArCH),132.7(ArCH),133.6(ArCH),133.9(季ArC),135.5(季ArC),135.6(季ArC),136.0(季ArC),140.3(季ArC),171.2,174.1,180.1(CN和2xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表16和图15。[表16](实施例4)手性甘氨酸等价物与羰基化合物的羟醛缩合反应和光学活性氨基酸的合成实施例4-1:利用手性甘氨酸等价物与苯甲醛的羟醛缩合反应进行的在部分结构中具有D-苏式(threo)-3-苯基丝氨酸的Ni(II)络合物的合成在手性甘氨酸等价物(R体)(500mg,0.734mmol)的甲醇溶液(25mL)中添加苯甲醛(0.38mL,3.67mmol),在-5℃以下搅拌10分钟。然后,滴加DBU(0.33mL,2.20mmol),在-5℃以下搅拌2小时。将反应液加入到冰冷却的5%乙酸水溶液(25mL)中,搅拌30分钟,滤取析出的结晶。将该结晶在50℃下送风干燥,得到红色固体[555mg,82%de,(含有8.7%的手性甘氨酸等价物(R体))]。将所得到的红色固体利用硅胶柱层析[二氯甲烷:丙酮=97:3(v/v)]进行纯化后,进行再结晶,以红色结晶的形式得到在部分结构中具有D-苏式-3-苯基丝氨酸的Ni(II)络合物(150mg,收率26%)。ESI-MS(正离子模式):m/z=786.2,为[M+H]+1H-NMR(200MHz,CDCl3):δ2.27[1H,d,J=12.3Hz,一个氮杂C(α)H2N],2.88[1H,d,J=15.7Hz,一个氮杂C(α’)H2N],3.23[1H,dd,J=15.7,1.3Hz,一个氮杂C(α’)H2N],3.39[1H,d,J=12.3Hz,一个氮杂C(α)H2N],3.81(1H,d,J=13.4Hz,一个乙酰苯胺NCOCH2),4.20(1H,dd,J=13.4,1.3Hz,一个乙酰苯胺NCOCH2),4.28(1H,d,J=5.5Hz,AA部分的α-H),4.62(1H,dd,J=9.7,5.5Hz,AA部分的β-H),5.09(1H,d,J=9.7Hz,OH),6.70(1H,d,J=2.6Hz),7.03-7.10(1H,m,ArH),7.16-7.82(18H,m,ArH),7.87-8.00(4H,m,ArH),8.63(1H,d,J=9.3Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ57.7(CH2),58.9(CH2),61.5(CH2),73.2(CH),73.4(CH),124.9(ArCH),125.7(ArCH),126.1(ArCH),126.4(ArCH),126.6(ArCH),126.7(ArCH),127.2(ArCH),127.5(ArCH),127.7(ArCH),127.9(季ArC),128.15(ArCH),128.22(ArCH),128.3(ArCH),128.7(ArCH),128.9(ArCH),129.1(ArCH),129.7(ArCH),130.8(ArCH),130.9(季ArC),131.2(ArCH),131.4(季ArC),132.6(季ArC),132.7(ArCH),133.1(ArCH),133.6(季ArC),133.7(季ArC),134.6(季ArC),135.7(季ArC),140.2(季ArC),141.4(季ArC),171.6,174.0,177.6(CN和2xCO).实施例4-2:在部分结构中具有D-苏式-3-苯基丝氨酸的Ni(II)络合物在酸条件下的D-苏式-3-苯基丝氨酸的合成向在部分结构中具有D-苏式-3-苯基丝氨酸的Ni(II)络合物(100mg,0.127mmol)的甲醇(3mL)悬浊液中添加1N盐酸(0.64mL,0.635mmol),在50℃下搅拌4小时。将反应液在减压下浓缩,在残留物中加入二氯甲烷(10mL)和水(10mL)进行分层。分取水层,蒸馏除去溶剂后,将所得到的固体溶解到9%氨水(3mL)中,通入到阳离子交换树脂柱[SK-1B,18mL,洗脱液:2~4%氨水]中,得到D-苏式-苯基丝氨酸(15mg,0.083mmol,收率65%,99.4%ee)。另一方面,将有机层用4%氨水(10mL)、水(10mL)洗涤,接着用饱和盐水(10mL)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(R体)(49mg,0.086mmol,收率68%)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表17和图16。<HPLC条件-:D-苏式-3-苯基丝氨酸手性分析条件>柱:TSKgelEnantioL1(5μm,250×4.6mmi.d.)洗脱液:2mM硫酸铜水溶液(分析时间:15分钟)流量:0.8mL/分钟温度:35℃检测器:UV230nm[表17](实施例5)手性甘氨酸等价物与α,β-不饱和羰基化合物的迈克尔反应和光学活性氨基酸的合成实施例5-1:利用手性甘氨酸等价物与丙烯酸甲酯的迈克尔反应进行的在部分结构中具有L-谷氨酸-γ-甲基酯的Ni(II)络合物的合成在手性甘氨酸等价物(R体)(0.154g,0.226mmol)的甲醇悬浊液(3mL)中添加丙烯酸甲酯(0.029g,0.339mmol)、无水碳酸钾(0.005g,0.034mmol),在室温下搅拌2小时。将反应液加入到冰冷却的5%乙酸水溶液(30mL)中,搅拌30分钟,滤取析出的结晶。将滤取的结晶在50℃下送风干燥,以红色结晶的形式得到在部分结构中具有L-谷氨酸γ-甲基酯的Ni(II)络合物(0.164g,收率95.1%,93%de)。进而,将该结晶从二氯甲烷-甲醇中进行再结晶,得到在部分结构中具有L-谷氨酸γ-甲基酯的Ni(II)络合物(0.120mg,收率69.3%,96.0%de)。ESI-MS(正离子模式):m/z=766.4,为[M+H]+1H-NMR(200MHz,CDCl3):δ1.70-1.91(1H,m),2.11-2.30(1H,m),2.61-2.78(1H,m,Glu部分的一个γ-CH2),2.65[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.01[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.48(1H,ddd,J=16.8,9.5,5.7Hz,Glu部分的一个γ-CH2),3.60(3H,s,OMe),3.79和3.91(各1H,ABq,J=13.8Hz,乙酰苯胺NCOCH2),3.91(1H,dd,J=6.4,3.5Hz,Glu部分的α-H),4.67[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.80[1H,d,J=12.2Hz,一个氮杂C(α)H2N],6.60(1H,d,J=2.6Hz,ArH),7.04-7.17(2H,m,ArH),7.20-7.32(3H,m,ArH),7.35-7.57(7H,m,ArH),7.61(1H,d,J=8.2Hz,ArH),7.94-8.04(3H,m,ArH),8.16(1H,d,J=8.2Hz,ArH),8.47(1H,d,J=9.2Hz,ArH),8.82(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ28.0(CH2),30.2(CH2),52.2(OMe),58.7(NCOCH2),61.8和66.4(氮杂的2xCH2),70.1(Glu部分的α-CH),125.1(ArCH),126.1(季ArC),126.4(ArCH),126.7(ArCH),127.4(ArCH),127.76(ArCH),127.84(ArCH),128.1(季ArC),128.4(ArCH),128.6(ArCH),128.8(季ArC),129.1(ArCH),129.3(ArCH),129.4(ArCH),130.2(ArCH),131.0(季ArC),131.2(季ArC),131.5(季ArC),132.5(ArCH),132.7(ArCH),132.9(季ArC),133.7(季ArC),134.0(季ArC),135.4(季ArC),136.1(季ArC),141.1(季ArC),171.3,173.4,174.9,177.8(CN和3xCO).使用上述的<HPLC条件-1:络合物分析条件>,实施所得到的化合物的HPLC分析。将结果示于表18和图17。[表18]实施例5-2:在部分结构中具有L-谷氨酸γ-甲基酯的Ni(II)络合物在酸条件下的L-谷氨酸-γ-甲基酯的游离和利用Z基的保护向在部分结构中具有L-谷氨酸γ-甲基酯的Ni(II)络合物(0.12g,0.16mmol)的甲醇悬浊液(3.6mL)中添加6N盐酸(0.13mL,5eq.),在30℃~40℃下搅拌7小时。将反应液在减压下浓缩,使残留物溶解到二氯甲烷(4mL)中,将有机层用水(1mL)洗涤,将水层用二氯甲烷(2mL,2次)萃取。使用二氯甲烷使全部有机层为约20mL后,用饱和碳酸氢钠水溶液(5mL)、水(5mL)、饱和盐水(5mL)洗涤。将所得到的有机层用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(R体)(0.09g,收率98%)。另一方面,在水层中加入EDTA二氢二钠二水合物(0.06g,1eq.)、丙酮(1mL)和N-苄氧羰基氧基琥珀酰亚胺(0.16g,5eq.)的丙酮溶液(1mL),使用碳酸氢钠将pH调节为7-8后,搅拌过夜。在从反应液中减压蒸馏除去丙酮后的残留物中加入二氯甲烷(20mL),使用4N盐酸将pH调节为3,进行分层后,将水层用二氯甲烷(20mL)萃取。将全部有机层用饱和盐水(2mL)洗涤,用硫酸钠干燥后,浓缩干固,得到无色油状物质(0.16g)。将所得到的油状物质溶解到异丙醇(0.1mL)-乙酸乙酯(1mL)中,加入二环己胺(0.08g,3eq.)、石油醚(3mL)、己烷(3mL),搅拌过夜。滤取析出的结晶,在50℃下减压干燥,得到Z-L-谷氨酸-γ-甲基酯·DCHA盐(0.04g,收率56%,99.8%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表19和图18。<HPLC条件:Z-Glu(5-OMe)手性分析条件>柱:CHIRALCELOJ-RH(5μm,150×4.6mmi.d.)洗脱液:A:B=75:25(0~20分钟)A=0.1%磷酸水溶液B=0.1%磷酸乙腈溶液流量:1.0mL/分钟温度:30℃检测器:UV220nm[表19]实施例5-3:利用手性甘氨酸等价物与丙烯酰胺的迈克尔反应进行的在部分结构中具有L-谷氨酰胺的Ni(II)络合物的合成在氩气气氛下,在手性甘氨酸等价物(R体)(0.100g,0.147mmol)的甲醇悬浊液(1mL,甲醇预先脱气)中添加丙烯酰胺(0.016g,0.220mmol)、无水碳酸钾(0.060g,0.441mmol),在45℃下搅拌2小时。在反应液中加入冰冷却的5%乙酸水溶液(5mL)、二氯甲烷(5mL)并搅拌后,进行分层。将水层用二氯甲烷(5mL)萃取后,将全部有机层用水(2.5mL,5次)、饱和盐水(2.5mL)洗涤,用硫酸钠干燥后,浓缩干固。将浓缩残留物利用硅胶层析(二氯甲烷:甲醇=95:5)进行纯化,以红色结晶的形式得到在部分结构中具有L-谷氨酰胺的Ni(II)络合物(0.096g,收率86.7%,99.8%de)。ESI-MS(正离子模式):m/z=751.3,为[M+H]+1H-NMR(200MHz,CDCl3):δ1.68-1.88(1H,m),2.09-2.25(1H,m),2.34-2.70(2H,m),2.72[1H,d,J=12.2Hz,一个氮杂C(α)H2N],3.00[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.62和3.73(各1H,ABq,J=13.7Hz,乙酰苯胺NCOCH2),3.79(1H,dd,J=8.7,4.3Hz,Gln部分的α-H),4.56[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.84[1H,d,J=12.2Hz,一个氮杂C(α)H2N],5.20(1H,brs,一个CONH2),6.38(1H,brs,一个CONH2),6.66(1H,d,J=2.4Hz,ArH),6.94-7.01(1H,m,ArH),7.13-7.20(1H,m,ArH),7.21-7.33(3H,m,ArH),7.37-7.59(8H,m,ArH),7.86-8.01(3H,m,ArH),8.15(1H,d,J=8.2Hz,ArH),8.45(1H,d,J=9.2Hz,ArH),8.74(1H,d,J=8.4Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ30.2(CH2),31.2(CH2),58.4(NCOCH2),61.9和66.2(氮杂的2xCH2),69.8(Gln部分的α-CH),125.2(ArCH),126.1(季ArC),126.5(ArCH),126.6(ArCH),127.3(ArCH),127.5(ArCH),127.8(ArCH),128.0(ArCH),128.1(季ArC),128.4(ArCH),128.6(ArCH),128.8(季ArC),129.0(ArCH),129.1(ArCH),129.3(ArCH),129.5(ArCH),130.3(ArCH),131.1(季ArC),131.2(季ArC),131.4(季ArC),132.6(ArCH),132.7(ArCH),133.6(季ArC),133.9(季ArC),135.5(季ArC),136.1(季ArC),141.0(季ArC),170.7,173.6,174.8,178.5(CN和3xCO).利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表20和图19。<HPLC条件-2:络合物分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=40:60(0~40分钟)10:90(40分钟~50分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:0.5mL/分钟温度:30℃检测器:UV254nm[表20]实施例5-4:在部分结构中具有L-谷氨酰胺的Ni(II)络合物在酸条件下的L-谷氨酰胺的游离和利用Z基的保护向在部分结构中具有L-谷氨酰胺的Ni(II)络合物(0.10g,0.13mmol)的甲醇悬浊液(2.9mL)中添加1N盐酸(0.6mL,5eq.),在30℃下搅拌2小时。将反应液在减压下浓缩,使残留物溶解到二氯甲烷(5mL)中,将有机层用水(2.5mL)洗涤,将水层用二氯甲烷(5mL)萃取。将全部有机层用饱和碳酸氢钠水溶液(2.5mL)、水(2.5mL)、饱和盐水(2.5mL)洗涤,用硫酸钠干燥后,浓缩干固,回收手性助剂(R体)(0.07g,收率95%)。另一方面,在水层中添加EDTA二氢二钠二水合物(0.054g,1.1eq.)、丙酮(1mL),加入N-苄氧羰基氧基琥珀酰亚胺(0.043g,1.1eq.)的丙酮溶液(1.5mL),用1N氢氧化钠水溶液将pH调节为9后,搅拌2.5小时。在从反应液中减压蒸馏除去丙酮后的残留物中加入乙酸乙酯(8mL)和1N盐酸(1mL),进行分层后,将水层用乙酸乙酯(5mL,2次)萃取。将全部有机层用饱和盐水(2mL)洗涤,用硫酸钠干燥后,浓缩干固,得到褐色油状物质(0.05g)。将所得到的油状物质溶解到异丙醇(0.07mL)、乙酸乙酯(1mL)中,加入二环己胺(0.03g,1eq.),搅拌过夜。滤取析出的结晶,在50℃下减压干燥,得到Z-L-谷氨酰胺·DCHA盐(0.03g,收率48%,99.2%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表21和图20。<HPLC条件:Z-Gln手性分析条件>柱:CHIROBIOTICT(5μm,150×4.6mmi.d.)洗脱液:A:B=30:70A=甲醇B=0.1%三乙基乙酸铵缓冲液(pH4.1)流量:0.7mL/分钟温度:40℃检测器:UV254nm[表21]实施例5-5:利用手性甘氨酸等价物与巴豆酸甲酯的迈克尔反应进行的在部分结构中具有3-甲基-L-谷氨酸-γ-甲基酯的Ni(II)络合物的合成在氩气气氛下,在手性甘氨酸等价物(R体)(0.400g,0.588mmol)的甲醇悬浊液(8mL,甲醇预先脱气)中添加巴豆酸甲酯(0.130g,1.175mmol)、无水碳酸钾(0.243g,1.763mmol),在室温下搅拌2小时。在反应液中加入冰冷却的5%乙酸水溶液(5mL)、二氯甲烷(10mL)并搅拌后,进行分层。将有机层用水(5mL,2次)、饱和盐水(5mL)洗涤,用硫酸钠干燥后,浓缩干固,将所得到的浓缩残留物(469.0mg,目标物:目标物的异构体1:目标物的异构体2=83.2:13.8:2.9)利用硅胶层析(苯:丙酮=95:5)进行纯化,以红色结晶的形式得到在部分结构中具有3-甲基-L-谷氨酸γ-甲基酯的Ni(II)络合物(0.300g,收率65.4%,化学纯度>99%)。使用上述的<HPLC条件-1:络合物分析条件>实施纯化前的化合物的HPLC分析。将结果示于表22和图21。[表22]需要说明的是,本实施例中,目标物是α碳的立体构型为S构型且β碳的立体构型为S构型的化合物,目标物的异构体1是α碳为S构型且β碳为R构型的化合物。另外,目标物的异构体2是α碳为R构型且β碳的立体构型为S构型的化合物。ESI-MS(正离子模式):m/z=779.9,为[M+H]+1H-NMR(200MHz,CDCl3):δ1.97-2.32(6H,m),2.65[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.06[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.49(3H,s,OMe),3.64和3.73(各1H,ABq,J=13.9Hz,乙酰苯胺NCOCH2),3.84(1H,d,J=2.4Hz,氨基酸部分的α-H),4.55[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.76[1H,d,J=12.1Hz,一个氮杂C(α)H2N],6.62(1H,brd,J=2.6Hz,ArH),6.95-7.02(1H,m,ArH),7.09(1H,brd,J=7.0Hz,ArH),7.20-7.31(3H,m,ArH),7.34-7.59(8H,m,ArH),7.94-8.03(3H,m,ArH),8.16(1H,d,J=8.2Hz,ArH),8.46(1H,d,J=9.2Hz,ArH),8.82(1H,d,J=8.4Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ16.3(3-Me),35.1(β-CH),37.9(γ-CH2),51.5(OMe),59.1(NCOCH2),61.6和66.6(氮杂的2xCH2),74.1(α-CH),125.0(ArCH),126.1(季ArC),126.3(ArCH),126.4(ArCH),126.9(ArCH),127.2(ArCH),127.4(ArCH),127.5(ArCH),127.9(ArCH),128.3(季ArC),128.4(ArCH),128.6(ArCH),129.1(ArCH),129.35(ArCH),129.42(ArCH),130.3(ArCH),130.9(季ArC),131.2(季ArC),131.5(季ArC),132.6(ArCH),133.7(ArCH),134.0(季ArC),135.5(季ArC),136.0(季ArC),141.1(季ArC),170.7,172.0,174.4,176.1(CN和3xCO).实施例5-6:在部分结构中具有3-甲基-L-谷氨酸-γ-甲基酯的Ni(II)络合物在酸条件下的(2S,3S)-3-甲基-L-谷氨酸-γ-甲基酯的游离和利用Z基的保护、以及(2S,3S)-3-甲基-L-谷氨酸的立体结构的确定向在部分结构中具有(2S,3S)-3-甲基-L-谷氨酸γ-甲基酯的Ni(II)络合物(0.30g,0.384mmol)的甲醇悬浊液(9mL)中添加1N盐酸(2.3mL,6eq.),在50℃下搅拌4小时。将反应液在减压下浓缩,将所得到的残留物溶解到乙酸乙酯(6mL)中,将其用水(2mL)洗涤后,将水层用乙酸乙酯(1mL)萃取。将全部有机层用饱和碳酸氢钠水溶液(2mL)、水(2mL)、饱和盐水(2mL)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(0.22g,定量性的)。另一方面,在水层中加入EDTA二氢二钠二水合物(0.14g,1eq.)、丙酮(1mL)、水(1.1mL),添加N-苄氧羰基氧基琥珀酰亚胺(0.12g,1.2eq.)的丙酮溶液(1mL),加入碳酸氢钠将pH调节为7-8后,搅拌过夜。在从反应液中蒸馏除去丙酮后的残留液中加入乙酸乙酯(5mL)和1N盐酸并搅拌后,将水层的pH调节为2-3,进行分层后,将水层用乙酸乙酯(5mL和2.5mL)萃取。将全部有机层用饱和盐水(1mL)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,得到黄色油状物质(0.16g)。将该油状物质溶解到水(1mL)和甲醇(2mL)中,加入氢氧化钾(0.10g,4eq.),在室温下搅拌4.5小时。在从反应液中蒸馏除去甲醇后的残留液中加入水(20mL)、乙酸乙酯(2mL)和己烷(1mL),进行分层。将水层用乙酸乙酯(2mL,2次)洗涤后,使用4N盐酸将pH调节为6.5后,将水层进一步用乙酸乙酯(2mL,3次,1mL,6次)洗涤。接着,使用4N盐酸将pH调节为2后,将水层用乙酸乙酯(20mL和10mL)萃取。将全部有机层用饱和盐水(1.5mL)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,以无色油状物质的形式得到(2S,3S)-Z-3-甲基-L-谷氨酸(0.074g,收率68.4%,化学纯度97.5%)。在如上得到的(2S,3S)-Z-3-甲基-L-谷氨酸(0.04g,0.14mmol)的甲醇溶液(1mL)中加入10%Pd/C(0.002mg,0.5mol%),在氢气气氛下在室温下搅拌1小时。滤去Pd/C后,蒸馏除去溶剂,以白色固体的形式得到(2S,3S)-3-甲基-L-谷氨酸(0.02g,收率98.6%)。[α]25D=+40.8°(c0.19,6NHCl)1H-NMR(200MHz,NaOD/D2O):δ3.01(1H,d,J=5.7Hz),2.34(2H,dd,J=3.3和13.0Hz),1.95-2.10(1H,m),1.84(1H,dd,J=11.3和12.8Hz),0.87(3H,d,J=6.4Hz)作为参考,以下示出文献值(M.Xianetal.,J.Org.Chem.,2007,72,7560)。(2S,3S)-3-甲基-L-谷氨酸[α]25D=+42.0°(c0.9,6NHCl)1H-NMR(400MHz,NaOD/D2O)δ2.96(1H,d,J=6.0Hz),2.26(2H,dd,J=4.0和13.5Hz),1.96(1H,m),1.79(1H,dd,J=11.0和13.0Hz),0.81(3H,d,J=7.0Hz)(2S,3R)-3-甲基-L-谷氨酸[α]25D=+18.2°(c0.9,6NHCl)1H-NMR(400MHz,NaOD/D2O)δ3.14(1H,d,J=4.0Hz),2.22(2H,dd,J=5.0和13.0Hz),2.18(1H,m),1.98(1H,dd,J=9.5和13.0Hz),0.79(3H,d,J=7.0Hz)实施例6:在部分结构中具有(S)-α-烯丙基丙氨酸的Ni(II)络合物在酸条件下的(S)-α-烯丙基丙氨酸的游离在实施例3-4中合成的在部分结构中具有(S)-α-烯丙基丙氨酸的Ni(II)络合物(300.0mg,0.408mmol)的甲醇(9.0mL)悬浊液中添加1N盐酸(2.0mL,2.04mmol),在40℃下搅拌1小时。将反应液在减压下浓缩,在残留物中加入二氯甲烷(20mL)和水(20mL),溶解后分层。将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(R体)(255mg,收率为定量性的)。另一方面,将水层浓缩干固,将所得到的固体溶解到水(7.0mL)、1N盐酸(1.0mL)和甲醇(2.0mL)的混合溶剂中,通入到阳离子交换树脂柱(SK-1B,10mL,洗脱液:水、接着为2%氨水)中,以白色固体的形式得到(S)-α-烯丙基丙氨酸(47.7mg,0.369mmol,收率90%,>98%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表23和图22。<HPLC条件:(S)-α-烯丙基丙氨酸手性分析条件>柱:CHIROBIOTICT(5μm,250×4.6mmi.d.)洗脱液:A:B=30:70A=水B=乙醇流量:0.5mL/分钟温度:40℃检测器:UV210nm[表23]实施例7-1:利用在部分结构中具有丙氨酸的Ni(II)络合物与苄基氯甲基硫醚的反应进行的在部分结构中具有(R)-S-苄基-α-甲基半胱氨酸的Ni(II)络合物的合成在氩气气氛下,向在部分结构中具有丙氨酸的Ni(II)络合物(1.0g,1.44mmol)的二甲基甲酰胺(DMF)溶液(3mL)中加入氢氧化钠(288mg,7.20mmol),在-20℃下搅拌5分钟后,依次加入苄基氯甲基硫醚(621mg,3.60mmol)和碘化钾(657mg,3.96mmol),在0℃下搅拌1小时。将反应液加入到冰冷却的5%乙酸水溶液(40mL)中,过滤出析出的结晶。将滤取的结晶利用硅胶柱层析(二氯甲烷:丙酮=40:1)进行纯化,以红色结晶的形式得到在部分结构中具有(R)-S-苄基-α-甲基半胱氨酸的Ni(II)络合物(585mg,收率48.9%,>99.9%de)。ESI-MS(正离子模式):C48H38ClN3NiO3S[M+H]+的m/z计算值830.18;实测值830.11H-NMR(200MHz,CDCl3):δ1.00(3H,s,α-Me),2.67和2.83(各1H,ABq,J=12.0Hz,氨基酸部分的β-CH2),2.75[1H,d,J=12.0Hz,一个氮杂C(α)H2N],3.08[1H,d,J=15.1Hz,一个氮杂C(α’)H2N],3.64和3.83(各1H,ABq,J=13.8Hz,乙酰苯胺NCOCH2),3.98和4.07(各1H,ABq,J=13.1Hz,SCH2Ph),4.43[1H,d,J=15.1Hz,一个氮杂C(α’)H2N],4.80[1H,d,J=12.0Hz,一个氮杂C(α)H2N],6.66-6.73(1H,m,ArH),6.72(1H,d,J=2.6Hz),7.16-7.56(17H,m,ArH),7.91-8.01(3H,m,ArH),8.11(1H,d,J=8.4Hz,ArH),8.31(1H,d,J=9.0Hz,ArH),8.67(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ28.4(Me),38.1(CH2),42.3(CH2),58.9(NCOCH2),61.5和66.1(氮杂的2xCH2),与CDCl3的信号重叠的氨基酸部分的一个季α-碳原子,125.0(ArCH),125.9(季ArC),126.2(ArCH),126.3(ArCH),126.6(ArCH),127.3(ArCH),127.4(ArCH),127.5(ArCH),128.1(ArCH),128.4(ArCH),128.5(ArCH),128.6(季ArC),128.7(ArCH),128.9(季ArC),129.0(ArCH),129.1(ArCH),129.2(ArCH),129.5(ArCH),129.8(ArCH),130.4(季ArC),131.2(季ArC),131.3(季ArC),131.4(季ArC),132.2(ArCH),132.6(ArCH),133.6(季ArC),133.9(季ArC),135.3(季ArC),135.5(季ArC),136.0(季ArC),137.7(季ArC),140.5(季ArC),171.6,174.3,179.7(CN和2xCO).利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表24和图23。<HPLC条件:络合物分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=40:60~0:100(0~50分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:1.0mL/分钟温度:30℃检测器:UV254nm[表24]实施例7-2在部分结构中具有(R)-S-苄基-α-甲基半胱氨酸的Ni(II)络合物在酸条件下的(R)-S-苄基-α-甲基半胱氨酸的游离向在部分结构中具有(R)-S-苄基-α-甲基半胱氨酸的Ni(II)络合物(435mg,0.523mmol)的甲醇(13mL)悬浊液中添加1N盐酸(3.0mL,2.62mmol),在40-50℃下搅拌1小时。将反应液在减压下浓缩,在残留物中加入二氯甲烷(20mL)和水(20mL)进行分层。将有机层用2%氨水(10mL,2次)、水(10mL,2次)洗涤,接着用饱和盐水(10mL,2次)洗涤,用硫酸钠干燥后,蒸馏除去溶剂,回收手性助剂(R体)(281mg,收率95%)。另一方面,将水层浓缩干固,将所得到的固体溶解到水(5.0mL)、1N盐酸(2.0mL)和甲醇(4.0mL)的混合溶剂中,通入到阳离子交换树脂柱(SK-1B,10mL,洗脱液:水、接着为2%氨水)中,以浅黄色固体的形式得到(R)-S-苄基-α-甲基半胱氨酸(52mg,0.230mmol,收率44%,>98%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表25和图24。<HPLC条件:(R)-S-苄基-α-甲基半胱氨酸手性分析条件>柱:CHIROBIOTICT(5μm,250×4.6mmi.d.)洗脱液:A:B=50:50A=水B=乙醇流量:0.3mL/分钟温度:40℃检测器:UV210nm[表25]实施例8:利用在部分结构中具有丙氨酸的Ni(II)络合物与苄基氯甲基醚的反应进行的在部分结构中具有(S)-O-苄基-α-甲基丝氨酸的Ni(II)络合物的合成在氩气气氛下在-10~15℃下向在部分结构中具有丙氨酸的Ni(II)络合物(150mg,0.22mmol)的THF溶液(1.5mL)中滴加NaH(0.062g,1.295mmol),接着滴加苄基氯甲基醚(0.169g,1.079mmol)的THF溶液(1.5mL),在该温度下搅拌5小时。反应结束后,将反应液加入到冰冷却的5%乙酸铵水溶液(10mL)中,用二氯甲烷(10mL)萃取后,将所得到的有机层用水(20mL,2次)洗涤,接着用饱和盐水(20mL)洗涤。将有机层用硫酸钠干燥后,浓缩干固,将所得到的浓缩残留物利用硅胶柱层析(苯:丙酮=95:5)进行纯化,以红色结晶的形式得到在部分结构中具有(S)-O-苄基-α-甲基丝氨酸的Ni(II)络合物(70mg,收率40.0%,>99.9%de)。ESI-MS(正离子模式):C48H38ClN3NiO4[M+H]+的m/z计算值813.19;[M+MeOH+H]+的实测值846.11H-NMR(200MHz,CDCl3):δ0.74(3H,s,α-Me),2.77[1H,d,J=11.9Hz,一个氮杂C(α)H2N],3.21[1H,d,J=15.7Hz,一个氮杂C(α’)H2N],3.35和3.67(各1H,ABq,J=14.0Hz,乙酰苯胺NCOCH2),4.43和4.54(各1H,ABq,J=11.8Hz,氨基酸部分的β-CH2),4.45[1H,d,J=15.7Hz,一个氮杂C(α’)H2N],4.93[1H,d,J=11.9Hz,一个氮杂C(α)H2N],5.05(1H,d,J=6.0Hz,一个OCH2Ph),5.72(1H,d,J=6.0Hz,一个OCH2Ph),6.73(1H,d,J=2.6Hz),6.97-7.04(1H,m,ArH),7.15-7.57(17H,m,ArH),7.84(1H,d,J=8.1Hz,ArH),7.92-8.00(2H,m,ArH),8.04(1H,d,J=8.2Hz,ArH),8.65(1H,d,J=9.3Hz,ArH),8.81(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ20.5(Me),58.3(NCOCH2),61.7和66.1(氮杂的2xCH2),71.7(CH2),与CDCl3的信号重叠的氨基酸部分的一个季α-碳原子,89.5(CH2),124.9(ArCH),125.4(季ArC),126.0(ArCH),126.2(ArCH),126.3(ArCH),127.4(ArCH),127.5(ArCH),127.7(ArCH),127.9(ArCH),128.0(ArCH),128.2(ArCH),128.4(ArCH),128.5(ArCH),128.8(ArCH),129.0(ArCH),129.1(季ArC),129.2(季ArC),130.0(ArCH),131.2(季ArC),131.3(季ArC),131.7(季ArC),132.6(ArCH),133.6(季ArC),133.8(ArCH),133.9(季ArC),135.2(季ArC),135.6(季ArC),135.9(季ArC),136.7(季ArC),142.3(季ArC),168.5,170.5,174.6(CN和2xCO).利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表26和图25。<HPLC条件:络合物分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=40:60~0:100(0~50分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:1.0mL/分钟温度:30℃检测器:UV254nm[表26]实施例9-1:利用在部分结构中具有甘氨酸的Ni(II)络合物与反式-1,4-二溴-2-丁烯的反应进行的在部分结构中具有(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的Ni(II)络合物的合成在大气气氛下在甘氨酸等价物(S体)(33.0g,0.049mol)的二氯甲烷溶液(1320mL)中添加反式-1,4-二溴-2-丁烯(103.7g,0.485mol)、四丁基碘化铵(4.5g,0.012mol)和30%氢氧化钠水溶液(1320mL,9.9mol),在室温下搅拌30分钟。对反应溶液进行分层,将水层用二氯甲烷(800mL)萃取2次。将全部有机层用水(900mL)洗涤2次,用硫酸钠(350g)干燥后,将滤液浓缩,得到橙红色固体(162.9g)。将所得到的橙红色固体利用硅胶柱层析(二氯甲烷:丙酮=10:1)进行纯化,以红色结晶的形式得到在部分结构中具有4-溴-2-丁烯基甘氨酸的Ni(II)络合物(34.2g,收率86.7%,非对映异构体比(S-R:S-S)=70:30)。ESI-MS(正离子模式):C43H33BrClN3NiO3[M+H]+的m/z计算值812.08;实测值812.1主要非对映异构体的数据1H-NMR(200MHz,CDCl3,主要非对映异构体的数据):δ2.14-2.56(2H,m,β-CH2),2.70[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.03[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.69和3.81(各1H,ABq,J=14.0Hz,乙酰苯胺NCOCH2),4.23(2H,dd,J=7.0,0.9Hz,BrCH2),4.38-4.46(1H,m,α-CH),4.69[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.82[1H,d,J=12.1Hz,一个氮杂C(α)H2N],5.80-6.06(2H,m,2x烯烃的CH),6.66(1H,d,J=2.6Hz,ArH),6.91-6.99(1H,m,ArH),7.08-7.61(12H,m,ArH),7.92-8.02(3H,m,ArH),8.14(1H,d,J=8.2Hz,ArH),8.46(1H,d,J=9.2Hz,ArH),8.73(1H,d,J=8.2Hz,ArH).13C-NMR(50.3MHz,CDCl3,仅主要非对映异构体的主要信号):δ32.5(CH2),36.3(CH2),58.6(NCOCH2),61.9和66.4(氮杂的2xCH2),70.5(CH),170.5,174.7,177.6(CN和2xCO).接着,在氩气气氛下在0℃下向在部分结构中具有4-溴-2-丁烯基甘氨酸的Ni(II)络合物(53.0g,0.0651mol)的THF溶液(1060mL)中滴加2M叔丁醇钠THF溶液(48.8mL,0.098mol),在0℃下搅拌10分钟。在反应溶液中加入水(500mL)和二氯甲烷(500mL)进行分层,分取有机层后,将水层用二氯甲烷(500mL)萃取2次。将全部有机层用硫酸钠(270g)干燥后,将滤液浓缩干固,得到橙红色固体(51.5g)。将所得到的橙红色固体用乙酸乙酯(250mL,5v/w)进行2小时浆液洗涤后,将滤取的结晶用乙酸乙酯(100mL,2v/w)进一步洗涤,以红色结晶的形式得到在部分结构中具有(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的Ni(II)络合物(43.0g,收率90.0%,99.5%de)。ESI-MS(正离子模式):C43H32ClN3NiO3[M+H]+的m/z计算值732.16;实测值732.41H-NMR(200MHz,CDCl3):δ0.33(1H,dd,J=9.8,7.1Hz,环丙烷的一个CH2),1.48(1H,dd,J=9.3,7.1Hz,环丙烷的一个CH2),1.91-2.07(1H,m,环丙烷的CH),2.67[1H,d,J=12.1Hz,一个氮杂C(α)H2N],3.03[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],3.58和3.65(各1H,ABq,J=14.0Hz,乙酰苯胺NCOCH2),4.60[1H,d,J=15.6Hz,一个氮杂C(α’)H2N],4.69[1H,d,J=12.1Hz,一个氮杂C(α)H2N],5.27-5.34(1H,m,乙烯的CH),5.59-5.67(2H,m,乙烯的CH2),6.66(1H,d,J=2.4Hz,ArH),6.81-6.89(1H,m,ArH),7.12-7.19(1H,m,ArH),7.20-7.61(11H,m,ArH),7.93-8.02(3H,m,ArH),8.16(1H,d,J=8.2Hz,ArH),8.41(1H,d,J=9.2Hz,ArH),8.81(1H,d,J=8.4Hz,ArH).13C-NMR(50.3MHz,CDCl3):δ25.8(CH2),40.1(CH),58.8(NCOCH2),61.5和66.6(氮杂的2xCH2),来源于与CDCl3的信号重叠的氨基酸部分的一个季α-碳原子的信号,118.4(乙烯的CH2),124.5(ArCH),126.1(季ArC),126.3(季ArC),126.4(ArCH),127.1(ArCH),127.3(ArCH),127.5(ArCH),127.8(ArCH),128.4(ArCH),128.6(ArCH),128.7(ArCH),129.1(ArCH),129.4(ArCH),129.5(ArCH),130.6(ArCH),131.0(季ArC),131.2(季ArC),131.5(季ArC),132.6(ArCH),132.8(ArCH),133.7(季ArC),134.0(季ArC),134.6(ArCH),135.5(季ArC),136.0(季ArC),140.9(季ArC),165.2,173.2,174.3(CN和2xCO).利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表27和图26。<HPLC条件:络合物分析条件>柱:InertsilODS-3(3μm,150×4.6mmi.d.)洗脱液:A:B=40:60~0:100(0~50分钟)A=10mM甲酸铵0.1%甲酸缓冲液B=乙腈流量:1.0mL/分钟温度:30℃检测器:UV254nm[表27]<HPLC条件:络合物分析条件>实施例9-2:在部分结构中具有(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的Ni(II)络合物在酸条件下的(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的游离和利用Boc基的保护、以及(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的光学纯度的确定向在部分结构中具有(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸的Ni(II)络合物(32.0g,0.0437mol)的甲醇(960mL)悬浊液中添加1N盐酸(218.5mL,0.218mol),在50℃下搅拌1小时。将反应液的甲醇在减压下蒸馏除去,在残留物中加入水(300mL)和乙酸乙酯(300mL),进行分层。将水层用乙酸乙酯(200mL)洗涤后,浓缩干固,将所得到的固体溶解到水(200mL)、6NHCl(7mL)和甲醇(50mL)中,通入到阳离子交换树脂柱[SK-1B,200mL,洗脱液:2%氨水(800mL),4%氨水(1400mL)]中,得到(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸(4.47g,收率80.5%)。另一方面,将有机层用水(200mL)、2%氨水(100mL,2次)和饱和盐水(200mL)洗涤,用硫酸钠干燥后,浓缩干固,由此回收手性助剂(S体)(23.0g,收率92.8%)。接着,将通过上述操作得到的(1R,2S)-1-氨基-2-乙烯基环丙烷羧酸(4.47g,0.0352mol)溶解到水(100mL)和丙酮(100mL)中,添加(Boc)2O(8.4g,0.039mol)和三乙胺(3.9g,0.039mol),在室温下搅拌15小时。进而,添加(Boc)2O(3.8g,0.0175mol)和三乙胺(1.8g,0.0176mol),在室温下搅拌5小时。将反应液浓缩至100mL以下,加入柠檬酸(固体)而使水层的pH为2~3。将水层用乙酸乙酯(100mL,3次)萃取,将全部有机层用水(100mL)和饱和盐水(100mL)洗涤,用硫酸钠干燥后,浓缩滤液,以黄色油状物质的形式得到(1R,2S)-1-(Boc-氨基)-2-乙烯基环丙烷羧酸(8.6g,收率为定量性的)。将所得到的(1R,2S)-1-(Boc-氨基)-2-乙烯基环丙烷羧酸(8.6g)溶解到乙酸乙酯(34.4mL,4v/w)中,缓慢地加入二环己胺(6.4g,0.0352mol),在室温下搅拌20小时后,在0℃下搅拌1小时。滤取析出的结晶,用冷却至0℃的乙酸乙酯(34mL,4v/w)洗涤,以白色结晶的形式得到(1R,2S)-1-(Boc-氨基)-2-乙烯基环丙烷羧酸二环己胺盐(11.5g,两个步骤的收率80.0%)。1H-NMR(200MHz,CD3OD):δ1.10-1.49(10H,m),1.43(9H,s,tBu),1.61-2.16(13H,m),3.06-3.22(2H,m,2xCHN),4.92(1H,dd,J=10.3,2.2Hz),5.15(1H,dd,J=17.3,2.2Hz),5.88(1H,ddd,J=17.3,10.3,9.8Hz).13C-NMR(50.3MHz,CD3OD):δ23.1(3-CH2),25.7(DCHA的CH2),26.3(DCHA的CH2),29.0(Me3C),30.7(DCHA的CH2),33.4(2-CH),44.0(1-C,季),54.4(DCHA的CH),80.0(Me3C),115.3(CH=CH2),138.4(CH=CH2),158.1(CON),177.5(CO2H).使所得到的(1R,2S)-1-(Boc-氨基)-2-乙烯基环丙烷羧酸二环己胺盐(400mg,0.979mmol)悬浊到乙酸乙酯(4mL)中,在0℃下滴加5%乙酸水溶液(4mL),搅拌30分钟。在反应溶液中加入水(10mL)并进行分层,将水层用乙酸乙酯(10mL,3次)萃取。将全部有机层用水(10mL)洗涤2次,用硫酸钠干燥后,蒸馏除去溶剂,得到(1R,2S)-1-(Boc-氨基)-2-乙烯基环丙烷羧酸(236mg,收率为定量性的,99.6%ee)。1H-NMR(200MHz,CDCl3):δ1.24(1H,dd,J=9.3,4.9Hz,一个3-H2),1.37(9H,s,tBu),1.44-1.58(1H,m,一个3-H2),2.05(1H,dt,J=10.3,9.3Hz,2-H),5.04(1H,dd,J=10.3,2.2Hz),5.22(1H,dd,J=17.0,2.2Hz),5.68(1H,dt,J=17.0,10.3Hz),7.22(1Hx0.25,brs),7.57(1Hx0.75,brs),12.47(1H,brs).13C-NMR(50.3MHz,CDCl3):δ22.5(3-CH2,为主要旋转异构体),22.9(3-CH2,为次要旋转异构体),28.2(Me3C),32.5(2-CH,为主要旋转异构体),33.8(2-CH,为次要旋转异构体),40.6(1-C,季),78.0(Me3C),116.8(CH=CH2),135.0(CH=CH2),155.5(CON),172.5(CO2H,为主要旋转异构体),172.7(CO2H,为次要旋转异构体).利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表28和图27。<HPLC条件:手性分析条件>柱:CHIRALPAKAD-H(5μm,250×4.6mmi.d.)洗脱液:A:B=20:80(0~30分钟)A=异丙醇B=己烷流量:0.8mL/分钟温度:30℃检测器:UV220nm[表28]参考例2:在部分结构中具有L-苯丙氨酸的Ni(II)络合物在酸条件下的L-苯丙氨酸的游离和利用Z基的保护向在部分结构中具有L-苯丙氨酸的Ni(II)络合物(0.4g,0.52mmol)的甲醇悬浊液(12mL)中添加1N盐酸(2.6mL,5eq.),在40℃下搅拌6小时。反应结束后,将反应液浓缩,在残留物中加入二氯甲烷(10mL)使其溶解,将有机层用2%氨水(6mL,2次)、水(6mL,2次)萃取后,将有机层用饱和盐水(6mL,2次)洗涤。将所得到的有机层用硫酸钠干燥,滤去硫酸钠后,浓缩干固,以浅黄色固体的形式回收手性助剂(R体)(0.27g,收率90%)。将氨水层和水层萃取液合并,浓缩干固后,将所得到的固体溶解到9%氨水(3mL)中,通入到阳离子交换树脂柱[SK-1B,9mL,洗脱液:水、接着为氨水(2%→8%)]中,得到L-苯丙氨酸(0.083g,粗产物)。在L-苯丙氨酸(0.078g)中加入碳酸氢钠(0.041mg,1eq.)-碳酸钠(0.103mg,2eq.)水溶液(3mL)、丙酮(1mL)使其溶解,在冰浴中加入N-苄氧羰基氧基琥珀酰亚胺(0.121g,1eq.)的丙酮溶液(1mL),在室温下搅拌3小时。在将反应液浓缩而得到的残留物中加入乙酸乙酯(18mL)和1N盐酸(2.5mL),分层后,将水层用乙酸乙酯(18mL)萃取。将有机层用饱和盐水(5mL,2次)洗涤,用硫酸钠干燥后,浓缩,得到黄色油状物质(0.182g)。将所得到的黄色油状物质溶解到异丙醇(0.08mL)-乙酸乙酯(0.8mL)中,加入二环己胺(0.094g,1eq.)的乙酸乙酯溶液(0.4mL),进而加入乙酸乙酯(2.0mL),在室温下搅拌9小时。滤取析出的结晶,在50℃下送风干燥,以白色结晶的形式得到Z-L-苯丙氨酸·DCHA盐(0.178g,收率76%,99.0%ee)。利用以下的条件实施所得到的化合物的HPLC分析。将结果示于表29和图28。<HPLC条件:Z-Phe手性分析条件>柱:CHIRALCELOJ-RH(5μm,150×4.6mmi.d.)洗脱液:A:B=65:35(0~30分钟)A=0.1%磷酸水溶液B=0.1%磷酸乙腈溶液流量:0.5mL/分钟温度:35℃检测器:UV254nm[表29]产业上的可利用性根据本发明,能够高收率且高对映选择性地制造具有所期望的手性的光学活性α-氨基酸。另外,根据本发明,还能够高收率且高对映选择性地并且简便地制造在药品开发等中的重要性日益提高的光学活性α,α-二取代α-氨基酸。此外,根据本发明,能够提供在上述光学活性α-氨基酸和α,α-二取代α-氨基酸的制造方法中有用的中间体。此外,根据本发明,能够提供由天然型光学活性α-氨基酸(L型)或任意比例的光学活性α-氨基酸混合物简便地制造非天然型光学活性α-氨基酸(D型)的方法。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1