一种化合物、液晶组合物及液晶显示器的制作方法
本发明涉及液晶显示领域,尤其涉及一种化合物、液晶组合物及液晶显示器。
背景技术:
:按照不同的显示方式,液晶显示器分为动态闪射型、扭曲型、超扭曲型和平面转换型。虽然不同类型液晶显示器要求液晶组合物具有不同特性,但也要求其具有下述共性:适当的光学各向异性、适当的旋转粘度和较快的响应速度。液晶组合物是由多种液晶化合物混合制成,因此液晶化合物单体的性能显得尤为重要。现有的液晶组合物由于清亮点较低,不能满足液晶显示器在高温状态下使用的要求,且存在使用温度范围较窄的问题。因此希望提供一种能提高液晶组合物清亮点的化合物。技术实现要素:本发明提供了一种液晶化合物解决了现有技术中液晶组合物清亮点不高的问题;提供了一种液晶组合物和液晶显示器解决了液晶显示器不能在高温状态下使用以及工作温度范围较窄的问题。根据本发明的一方面,提供了一种化合物,该化合物如式I所示:其中,R1和R2各自独立的选自氢原子或1~15个碳原子的烷基或烷氧基;A、B和C各自独立地选自下述基团:Z1和Z2各自独立地选自:单键、-CH=CH-、-C≡C-、-COO-、-OOC-、-CF2O-、-OCH2-、-CH2O-、-OCF2-、-CH2CH2-、-CF2CH2-、-CH2CF2-、-C2F4-、-(CH2)4-和-CF=CF-;a,b,c,d和e各自独立地选自0、1和2。可选地,根据本发明的化合物,所述烷基或烷氧基中的一个或多个-CH2-各自独立地被-CH=CH-、-C≡C-、-COO-、-OOC-、-O-或环丁烷替代。可选地,根据本发明的化合物,所述烷基或烷氧基中一个或多个氢各自独立地被氟和/或氯取代。可选地,根据本发明的化合物,所述烷基或烷氧基各自独立地被CN、OCN、OCF3、CHF2、OCHF2、SCN、NCS或SF5取代。根据本发明的另一方面,提供了一种液晶组合物,该液晶组合物至少包括一种根据本发明的化合物。可选地,根据本发明的液晶组合物,该液晶组合物包括以重量百分比计5-40%的根据本发明的化合物。可选地,根据本发明的液晶组合物,该液晶组合物包括以重量百分比计的15-30%本发明的化合物。根据本发明的另一方面,提供了一种液晶显示器,含有根据本发明的液晶组合物。本发明的有益效果如下:根据本发明的化合物引入了氮杂双环辛烷的刚性结构,因此使本发明化合物具有高清亮点,液晶组合物的熔点一般在-10℃以下,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;将本发明化合物应用到液晶组合物中,提高了液晶组合物的清亮点,也提高了液晶组合物的工作温度范围。将该液晶组合物应用于液晶显示器中,由于液晶组合物清亮点提高满足了液晶显示器在高温下工作的需求,也提高了液晶显示器工作的温度范围。附图说明图1为化合物e的核磁谱图;图2为化合物j的核磁谱图。具体实施方式具体实施方式仅为对本发明的说明,而不构成对本
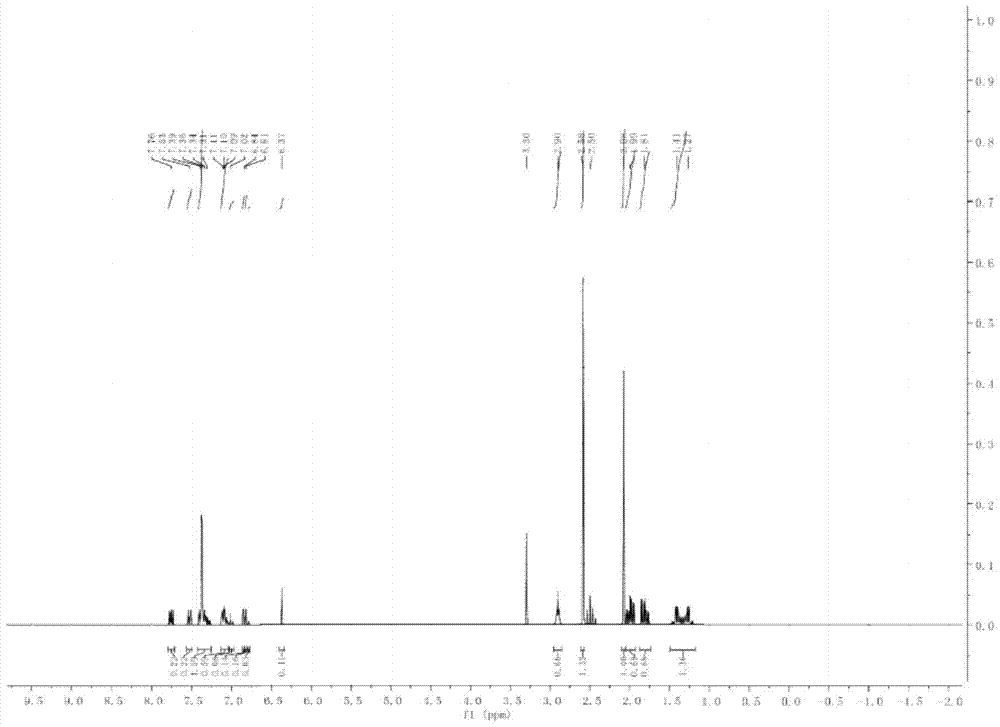
发明内容的限制,下面将结合附图和具体的实施方式对本发明进行进一步说明和描述。本发明提供了一种化合物,该化合物如式I所示:其中,R1和R2各自独立的选自氢原子或1~15个碳原子的烷基或烷氧基;A、B和C各自独立地选自下述基团:Z1和Z2各自独立地选自:单键、-CH=CH-、-C≡C-、-COO-、-OOC-、-CF2O-、-OCH2-、-CH2O-、-OCF2-、-CH2CH2-、-CF2CH2-、-CH2CF2-、-C2F4-、-(CH2)4-和-CF=CF-;a,b,c,d和e各自独立地选自0、1和2。根据本发明的化合物引入了氮杂双环辛烷刚性结构,使该化合物具有高清亮点,也提高了液晶组合物的工作温度范围;将该化合物应用到液晶组合物中,能提高液晶组合物的清亮点,也提高了液晶组合物的工作温度范围;将液晶组合物应用于液晶显示器中,满足了液晶显示器在高温下工作的需求,也扩大了液晶显示器工作的温度范围。根据本发明一种实施方式的化合物,烷基或烷氧基中的一个或多个-CH2-各自独立地被-CH=CH-、-C≡C-、-COO-、-OOC-、-O-或环丁烷替代。根据本发明一种实施方式的化合物,烷基或烷氧基中一个或多个氢各自独立地被氟和/或氯取代。根据本发明一种实施方式的化合物,烷基或烷氧基各自独立地被CN、OCN、OCF3、CHF2、OCHF2、SCN、NCS或SF5取代。根据本发明的化合物,优选表a中表示为I1-I12的化合物:表a在表a所示的化合物I1~I12中,R1为氢或碳原子数为1-10的直链烷基或烷氧基;(F)表示苯环上有氟原子取代基或为氢;R2为氢或碳原子数为1-10的直链烷基或烷氧基,也可以为Cl、F、CN、OCF3、CF3、SCN、CHF2、OCHF2。申请人对本申请式I所示化合物进行了合成及表征,在此列举两种具体化合物的合成方法及表征数据。在下述两种化合物的合成方法中,所用材料如无特别说明均从商业途径获得。其中在下述的实施方式和实施例中:GC表示气相色谱纯度;MS表示质谱;MP表示熔点;CP表示清亮点;△ε表示介电各向异性;△n表示光学各向异性;γ1表示旋转粘度;所得化合物用GC-MS所得质谱图和气相色谱来鉴定说明;GC-MS分析测定仪器为岛津公司的QP2010SE型;熔点使用的仪器为WRX-1S显微热分析仪,设定升温速率为3℃/min;清亮点采用了常规的测试方法。化合物的物性测定方法:1.光学各向异性利用阿贝折射计,在25℃、589nm波长条件下进行测定。在同一方向对主棱镜的表面进行摩擦,摩擦后将试样滴加到主棱镜上。折射率n11在偏光方向与摩擦方向平行时测定;折射率n⊥在偏光方向与摩擦方向垂直时测定;光学各向异性△n通过公式△n=n11-n⊥计算得出。2.介电常数各向异性使用惠普公司型号为HP4284a的仪器进行测定。在25℃,测定液晶分子在轴方向的介电常数∑11,以及液晶分子短轴方向的介电常数∑⊥,介电常数各向异性通过公式△∑=∑11-∑⊥计算得出。合成的具体化合物之一如式e所示:申请人对式e所示化合物的合成路线设计为:具体的合成步骤包括:步骤1:3-(3,5-二氟苯)-8-甲基-8-氮杂[3.2.1]双环辛烷-3-醇(化合物a)的合成。2L三口瓶中加入28.8g(1.2mol,1.2eq)新制镁屑和200mL干燥四氢呋喃、2粒碘作为引发剂,加热至回流,滴加19.3g(0.1mol)3,5-二氟溴苯引发,待引发格氏反应后,保持反应轻微回流,继续滴加212.3g(1.1mol,共1.2eq)3,5-二氟溴苯溶于500mL干燥四氢呋喃溶剂中。完毕后保持回流30分钟,再滴加139.2g(1mol,1eq)托品酮溶于500mL干燥四氢呋喃溶剂中,保持反应轻微回流,滴加完毕后再保持回流3小时,降至室温,搅拌下将反应液倒入1000g冰水和3mol盐酸的混合液中进行水解,搅拌30分钟,分液,取上层有机相,用500mL甲苯将水相提取两次,合并有机相。用1L去离子水分3次洗涤有机相,用无水硫酸钠干燥后,真空水浴蒸馏除去溶剂,得260g无色液体,即为化合物a。步骤2:3-(3,5-二氟苯)-8-甲基-8-氮杂[3.2.1]双环辛烷-3-烯(化合物b)的合成;在2L三口瓶中加入步骤1制得的260g化合物a作为反应物、20g对甲苯磺酸作为脱水剂、1.2L甲苯作为溶剂,回流分水3小时后停止,降至室温,加入饱和碳酸氢钠溶液200mL,搅拌10分钟后分液,取上层有机相,用500mL去离子水分两次洗涤有机相,用无水硫酸钠干燥后直接通过10厘米硅胶柱进行柱层析,之后使用200mL甲苯洗脱,水浴真空蒸馏除净溶剂,再加入200mL甲苯和400mL乙醇的混合溶剂进行重结晶,室温析出,然后在冰箱内冷冻4小时,过滤得到207.1g(0.88mol)化合物b,其收率为88.0%,GC检测其纯度为99.2%。步骤3:3-(3,5-二氟苯)-8-甲基-8-氮杂[3.2.1]双环辛烷(化合物c)合成。向2L氢化釜中加入步骤2得到的207.1g(0.88mol)化合物c、加入600mL甲苯和300mL乙醇作为混合溶剂、10g钯碳作为催化剂,盖好氢化釜的釜盖后用氢气排空气5次,加氢至釜内压力3.5Mpa,升温至120℃,搅拌加氢反应8小时,反应完毕,降温至40℃以下,过滤除去钯碳,用300mL甲苯洗涤滤饼,将合并后的滤液旋蒸除去溶剂,得到淡黄色液体197.5g(0.83mol),即为化合物c,收率为94.6%,GC检测其纯度为99.5%。步骤4:3-(4-(溴二氟甲基))-3,5-二氟苯-8-甲基-8-氮杂[3.2.1]双环辛烷(化合物d)的合成。向2L三口瓶中加入197.5g(0.83mol,1eq)化合物c、600mL干燥四氢呋喃作为溶剂,通氮气保护,降温至-60℃,滴加364ml2.5M(0.91mol,1.09eq)正丁基锂,滴加过程控温-55℃~-60℃,滴毕,继续控温搅拌反应1小时。降温至-70℃,滴加261.2g(1.25mol,1.50eq)二氟二溴甲烷,滴加过程控温-65℃~-70℃,滴毕,继续控温搅拌反应30分钟,升温至室温,加入200mL浓盐酸调节PH值、500mL水进行水解,然后进行分液,用500mL乙酸乙酯提取水相两次,合并有机相并水洗至中性,蒸干溶剂得淡黄色液体212.7g(0.58mol),即为化合物d,GC检测其纯度为82.6%,收率为70.1%。步骤5:3-(4-(3,4,5-三氟苯基)二氟甲氧基)-3,5-二氟苯-8-甲基-8-氮杂[3.2.1]双环辛烷(化合物e)合成。在反应瓶中加入212.7g(0.58mol,1eq)化合物d,500mlDMSO作为溶剂,160.3g(1.16mol,2eq)无水碳酸钾,103.1g(0.70mol)3,4,5-三氟苯酚(反应物),搅拌加热至65~70℃反应2小时。降温至室温,过滤固体,并用300mL二氯甲烷冲洗滤饼,滤液加入1L水,搅拌、分液,用500mL二氯甲烷提取两次水层,水洗有机相至中性,蒸干溶剂。浓缩物溶于500mL甲苯中,过硅胶柱脱色,用300mL甲苯进行洗脱,收集洗脱液并蒸除溶剂,所得物用无水乙醇重结晶3次,得到白色晶体100.5g(0.23mol),即为化合物e,GC检测其纯度为99.5%,收率42.5%。申请人对得到的化合物e的具体参数进行了测定,测定结果如下:MP:68℃;CP:121℃;△ε:21.0;△n:0.18;MS:124.11(1.2%)、414.13(10.5%)、433.13(100.0%)、434.13(23.7%)、435.13(2.9%);图1为该化合物的核磁谱图,对该核磁谱图的解析为:1HNMR(400MHz,d6-DMSO):δ6.85(s,1H)、6.71(d,J=10.0Hz,4H)、6.57(d,J=14.1Hz,4H)、3.75(s,1H)、2.96(s,3H)、2.73(s,6H)、2.08(s,4H)、1.99(s,3H)、1.85~1.60(m,5H)、1.53~1.17(m,9H)、0.98(s,1H)。根据上述测定结果,可以看出:(1)清亮点CP较高;(2)清亮点CP和熔点MP之间的差值较大,为53℃;(3)△ε和△n在正常值范围内;(4)根据MS证明其分子量与化合物e应该具备的分子量一致,根据核磁谱图解析证明了制备得到的化合物即为结构式e所示的化合物。合成的具体化合物之二如式j所示:其合成路线为:具体合成步骤包括:步骤1:3-(4-溴-苯基)-8-甲基-8-氮杂-双环[3.2.1]辛-3-醇(化合物h)合成。在1L三口瓶中加入47.2g(0.2mol,1eq)对二溴苯、300mL四氢呋喃,开启搅拌,在氮气保护下降温至-75℃,然后滴加0.22mol(1.1eq)丁基锂,滴毕保持-75℃~-70℃搅拌半小时,然后滴加33.4g(0.24mol,1.2eq)托品酮和50mL四氢呋喃的混合溶液,滴毕后自然升温至-40℃,保温半小时。将反应液倒入200g冰水和0.5mol盐酸的混合液中水解,搅拌15分钟后分液,取上层有机相,用100mL乙酸乙酯对水相提取两次,合并有机相。用300mL去离子水分3次洗涤有机相,用无水硫酸钠干燥后,减压蒸馏除去溶剂,得50.3g化合物h,GC检测含量92.0%,收率85.2%。步骤2:3-(4-溴-苯基)-8-甲基-8-氮杂-双环[3.2.1]辛烷(化合物i)合成。在1L三口瓶中加入50g(0.17mol,1.0eq)步骤1制得的化合物h、300mL二氯甲烷,开始搅拌,降温至-75℃后滴加19.7g(0.17mol,1.0eq)三乙基硅烷,滴毕后再滴加24.2g三氟化硼乙醚,滴毕保持-75℃~-70℃搅拌反应半小时,然后升温至-30℃,加入200mL饱和碳酸氢钠水溶液水解,搅拌10分钟,静置分液;水相用50mL二氯甲烷提取两次,合并有机相,将有机相水洗至中性,在常压下过硅胶柱后浓缩干,用无水乙醇重结晶,得白色固体34.7g,即为化合物i,GC检测含量98.5%,收率72.8%。步骤3:8-甲基-3-(3,4,5,2'-四氟三联苯-4'-基)-8-氮杂-二环[3.2.1]辛烷(化合物j)的合成。在1L三口瓶中加入34g(0.12mol,1eq)化合物i、32.4g(0.12mol,1eq)3',4',5'-三氟-3-氟联苯硼酸、19.1g(0.18mol,1.5eq)碳酸钠、1g四(三苯基膦)钯(催化剂)、100mL甲苯(溶剂)、100mL乙醇、100mL水,加热回流反应8小时,然后将反应液倒入分液漏斗中静置分层,水层用50mL甲苯萃取两遍,合并甲苯相,用100mL水洗两遍。蒸干甲苯,用石油醚重结晶三遍,得到白色晶体35.7g,即为目标产物化合物j,GC检测其纯度为99.8%,收率为69.2%。申请人对合成的化合物j的具体参数进行了测定,测定结果如下:MP:96℃;CP:129℃;△ε:13.3;△n:0.25;MS:124.11(0.8%)、406.18(2.5%)、425.18(100.0%)、426.18(29.3%)、427.18(4.1%);图2示出了该化合物的核磁谱图,对该谱图的解析为:1HNMR(400MHz,d6-DMSO):δ7.76(s,1H)、7.53(s,1H)、7.35(dd,J=25.0,15.0Hz,7H)、7.14–6.97(m,3H)、6.82(d,J=11.2Hz,2H)、6.37(s,1H)、2.90(s,3H)、2.58(s,6H)、2.08(s,4H)、1.90(d,J=88.9Hz,6H)、1.34(d,J=71.0Hz,6H)。根据上述测定结果,可以看出:(1)清亮点CP较高;(2)清亮点CP和熔点CP之间的差值较大;(3)△ε和△n在正常值范围内;(4)根据MS证明其分子量与化合物e应该具备的分子量一致,根据核磁谱图解析证明了制备得到的化合物即为结构式e所示的化合物。根据本发明的另一方面,提供了一种液晶组合物,该液晶组合物至少包括一种本发明的化合物。根据本发明一种实施方式的液晶组合物,该液晶组合物包括以重量百分比计5-40%的根据本发明的化合物。根据本发明一种实施方式的液晶组合物,该液晶组合物包括以重量百分比计15-30%的根据本发明的化合物。根据本发明的液晶组合物,不仅具有适当的光学各向异性、低旋转粘度和较快的响应速度,而且具有高清亮点,不仅可以应用于液晶显示器中,且该液晶显示器可以在高温下使用,可使用的温度范围较广。根据本发明的另一方面,提供了一种液晶显示器,含有根据本发明的液晶组合物。根据本发明的液晶显示器,使用了本发明的化合物组成的液晶组合物,由于根据本发明的化合物具备较高的清亮点,使含有该化合物的液晶组合物具有较高的清亮点,在该液晶组合物应用于液晶显示器中时,该液晶显示器可以在高温条件下工作;本发明化合物的清亮点与熔点之间的差值较大,在温度小于熔点时,化合物为固态,在温度大于清亮点时,化合物为液态,只有在熔点和清亮点之间的范围内,化合物才能以液晶态存在,因此增大了化合物清亮点和熔点之间的差值,也就增加了液晶组合物的清亮点和熔点之间的差值,因此增大了应用该液晶组合物的液晶显示器的工作温度范围。综上可以看出,根据本发明的化合物、液晶组合物和液晶显示器可选因素较多,利用本发明的权利要求可以组合出不同的实施例,因此实施例仅为对本发明的说明,而不作为对本发明的限制。下面将结合关于液晶组合物的实施例以及相应的对比例对本发明进行说明。其中,实施例1-6均为根据本发明的组合物,对比例1~6则均为相应不含有根据本发明的化合物的组合物,其中实施例和对比例中单体组分均为重量份;并分别对相应实施例和对比例中的液晶组合物的相关参数进行了测定,具体数据参见下述实施例和对比例。实施例1和对比例1:实施例1和对比例1的液晶组合物的组分如表1所示:表1对实施例1和对比例1的液晶组合物进行了一系列的性能测试,测试结果如表1-1所示:表1-1性能参数实施例1对比例1Cp92℃85℃Δn0.1890.109Δε7.67.8γ17375根据表1-1的测试结果可以看出:在实施例1中的液晶组合物中加入了本发明的化合物,其与未加入本发明化合物的对比例1相比明显提高了液晶组合物的清亮点(Cp),液晶组合物的熔点一般在-10℃以下,对比例1和实施例1中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点即提高了液晶组合物的工作温度范围;另外加入本发明的化合物,对液晶组合物的旋转粘度(γ1)和介电常数(Δε)的影响不大,因此可以使液晶组合物保持正常应用。在实施例1中加入的化合物和其它单体相互作用还增加了液晶组合物的光学各向异性(Δn),因此有助于减少液晶层厚度以减少液晶盒厚度,从而进一步减少了液晶显示器的厚度。实施例2和对比例2实施例2和对比例2的液晶组合物的组分如表2所示:表2对实施例2和对比例2的液晶组合物进行了一系列的性能测试,测试结果如表2-2所示:表2-2性能参数实施例2对比例2Cp92℃85℃Δn0.1790.182Δε7.97.8γ19694根据表2-2的测试结果可以看出:实施例2中加入了本发明的化合物,与未加入本发明化合物的对比例2相比,提高了液晶组合物的清亮点(Cp),液晶组合物的熔点一般在-10℃以下,对比例2和实施例2中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;液晶组合物的光学各向异性(Δn)、介电各向异性(Δε)和旋转粘度(γ1)变化不大;因此根据本实施例的液晶组合物提高了清亮点(Cp)但不影响其它参数性能。实施例3和对比例3实施例3和对比例3的液晶组合物的组分如表3所示:表3对实施例3和对比例3的液晶组合物进行了一系列的性能测试,测试结果如表3-3所示:表3-3性能参数实施例3对比例3Cp85℃80℃Δn0.1790.182Δε9.67.8γ116096根据表3-3的测试结果可以看出:实施例3中加入了本发明的化合物,与未加入本发明化合物的对比例3相比,提高了液晶组合物的清亮点(Cp),液晶组合物的熔点一般在-10℃以下,对比例3和实施例3中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;液晶组合物的光学各向异性(Δn)变化不大;提高了介电各向异性(Δε)因此当其应用于液晶显示器时可以提高液晶显示器的反应速度;同时旋转粘度(γ1)有所提高,但仍在正常的使用范围内,且介电各向异性(Δε)和旋转粘度(γ1)在对显示器反应速度的影响中,介电各向异性(Δε)占主导地位,因此综合来看,将实施例3的组合物应用于液晶显示器时,仍然提高了液晶显示器的反应速度。实施例4和对比例4实施例4和对比例4的液晶组合物的组分如表4所示:表4对实施例4和对比例4的液晶组合物进行了一系列的性能测试,测试结果如表4-4所示:表4-4性能参数实施例4对比例4Cp95℃85.0℃Δn0.1100.109Δε7.47.8γ17275根据表4-4的测试结果可以看出:实施例4中加入了本发明的化合物,与未加入本发明化合物的对比例4相比,提高了液晶组合物的清亮点,液晶组合物的熔点一般在-10℃以下,对比例4和实施例4中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;液晶组合物的光学各向异性(Δn)、介电各向异性(Δε)和旋转粘度(γ1)变化不大。因此可以看出在加入本发明的化合物后可以提高液晶组合物的清亮点,但并未影响其它的性能参数。实施例5和对比例5实施例5和对比例5的液晶组合物的组分如表5所示:表5对实施例5和对比例5的液晶组合物进行了一系列的性能测试,测试结果如表5-5所示:表5-5性能参数实施例5对比例5Cp84℃75.3℃Δn0.1820.171Δε9.27.8γ111175根据表5-5的测试结果可以看出:实施例5中加入了本发明的化合物,与未加入本发明化合物的对比例5相比,提高了液晶组合物的清亮点,液晶组合物的熔点一般在-10℃以下,对比例5和实施例5中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;液晶组合物的光学各向异性(Δn)变化不大;提高了介电各向异性(Δε)因此当其应用于液晶显示器时可以提高液晶显示器的反应速度;同时旋转粘度(γ1)也得到了提高,但仍在正常使用范围内,且介电各向异性(Δε)和旋转粘度(γ1)在对显示器反应速度的影响中,介电各向异性(Δε)起主导作用,因此综合来看,将实施例5的组合物应用于液晶显示器时,仍然提高了液晶显示器的反应速度。实施例6和对比例6实施例6和对比例6的液晶组合物的组分如表6所示:表6对实施例6和对比例6的液晶组合物进行了一系列的性能测试,测试结果如表6-6所示:表6-6性能参数实施例6对比例6Cp92℃86℃Δn0.1790.182Δε7.97.8γ19694根据表6-6的测试结果可以看出:实施例6中加入了本发明的化合物,与未加入本发明化合物的对比例6相比,提高了液晶组合物的清亮点(CP),液晶组合物的熔点一般在-10℃以下,对比例6和实施例6中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点,即提高了液晶组合物的工作温度范围;液晶组合物的光学各向异性(Δn)、介电各向异性(Δε)和旋转粘度(γ1)变化不大。因此可以看出在加入本发明的化合物后可以提高液晶组合物的清亮点,且同时不影响其它参数性能。实施例1-6得到的液晶组合物与其相对应的对比例相比,清亮点均得到了提高,满足了液晶显示器在高温下使用的需求,同时液晶组合物的熔点一般在-10℃以下,对比例和实施例中液晶组合物的熔点均足够低,完全小于正常使用的温度范围,所以熔点对使用温度影响不大,因此提高了清亮点即提高了液晶组合物的工作温度范围。显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1