运行气相光气化装置的方法与流程
大量制备异氰酸酯并主要用作制备聚氨酯的起始物质。它们通常通过使用化学计算过量的光气使相应的胺与光气反应制备。胺与光气的反应可以在气相也可以在液相中进行。在气相中的工艺方案(通常被称作气相光气化)的特征在于选择反应条件以使至少反应组分胺、异氰酸酯和光气,优选所有反应物、产物和反应中间产物在所选条件下为气态。气相光气化的优点尤其包括降低的光气存在(Aufkommen)(所谓的光气“滞留”)、避免难以光气化的中间产物、提高的反应收率和较低能量要求,因为使用较少溶剂。本发明仅涉及气相光气化,尤其涉及启动气相光气化装置的顺利方法。
现有技术公开了通过在气相中使胺与光气反应而制备异氰酸酯的各种方法。良好工艺方案的重要因素是气相光气化的反应物的良好混合。EP-A-0 289 840描述了通过气相光气化制备二异氰酸酯,其中根据该发明的制备在没有活动件的柱形空间内在湍流中在200℃至600℃的温度下进行。
EP-A-0 570 799 涉及制备芳族二异氰酸酯的方法,其中相应的二胺与光气的反应在管式反应器中在高于二胺的沸点下在0.5至5秒的平均接触时间内进行。
EP-A-0 699 657描述在气相中制备芳族二异氰酸酯的方法,其中相应的二胺与光气的反应在包含两个区的反应器中进行,其中构成总反应器体积的大约20%至80%的第一区具有理想的混合,构成总反应器体积的80%至20%的第二区可以以活塞流为特征。第二反应区优选为管式反应器的形式。
管式反应器用于气相光气化的优化是许多申请的主题,如在EP-A-0 570 799中使用喷射混合器原理公开了其原理(Chemie-Ing.-Techn. 44 (1972),第1055页, 图10)。
根据EP-A-1 362 847的教导,经管式反应器的环形空间供应的反应物料流的均化和两种反应物料流在尽可能中心地进给到管式反应器中对反应区的稳定性和因此对整个气相反应具有巨大的正面影响。
如EP-A-1 555 258中所述,所用管式反应器的扩大也使得必须扩大混合喷嘴,其通常为光滑喷射喷嘴的形式。但是,光滑喷射喷嘴的直径扩大也由于所需的较大扩散长度而降低中心射流的混合速度并提高回混风险,这又导致形成聚合杂质和反应器上的固体附着物。根据EP-A-1 555 258的教导,当一个反应物料流经由另一反应物料流中同心安置的环形间隙高速喷射时,可以消除所述缺点。这能使用于混合的扩散长度小并使混合时间极短。该反应可以随后以高选择性进行以产生所需异氰酸酯。由此减轻聚合杂质的存在和附着物的形成。
根据EP-A-1 526 129的教导,中心喷嘴中的反应物料流的湍流增强对反应物的混合和因此对整个气相反应具有正面影响。由于更好的充分混合,形成副产物的倾向降低。
EP-A-1 449 826公开了通过相应的二胺的光气化制备二异氰酸酯的方法,其中将任选用惰性气体或用惰性溶剂的蒸气稀释的气态二胺和光气分开加热到200℃至600℃的温度并在管式反应器中混合和反应,其中在管式反应器内布置平行于管式反应器的轴排列的数量n > 2的喷嘴,其中经n个喷嘴将包含二胺的料流供入管式反应器并经剩余自由空间将光气料流供入管式反应器。
管式反应器用于气相光气化时的进一步发展是WO 2007/028715的主题。所用反应器具有混合装置和反应空间。根据WO 2007/028715的教导,该反应空间在前区包含混合空间,在此主要发生反应物气态光气和胺(任选与惰性介质混合)的混合,其通常伴随着反应的开始。根据WO 2007/028715的教导,在反应空间的后部,随后基本仅发生反应,并最多在微小程度上发生混合。优选地,在WO 2007/028715中公开的方法中,使用相对于流向旋转对称的反应空间,其可以在构造上基本分割成在流动方向上沿反应器纵轴的最多四个纵向区段(Längenabschnitt),其中这些纵向区段在流经的横截面面积的尺寸上不同。
WO 2008/055898公开了通过相应的胺在反应器中在气相中的光气化制备异氰酸酯的方法,其中,类似于WO 2007/028715,所用反应器具有混合装置和反应空间,该旋转对称的反应空间可以在构造上基本分割成在流动方向上沿反应器纵轴的最多四个纵向区段,其中这些纵向区段在流经的横截面面积的尺寸上不同。但是,与WO 2007/028715相比,不借助安装到管式反应器中的体积物件(Volumenkörper)而是借助反应器外壁的相应扩展或收缩实现流经的横截面面积的变化。
EP-A-1 275 639同样公开了,作为通过相应的胺与光气在气相中的光气化制备异氰酸酯的可能的工艺变体,使用下述反应器:其中反应空间在流动方向上在这两种反应物混合后具有流经的横截面面积的扩展。通过横截面面积的适当选择的扩展,可以使反应混合物在反应器长度上的流速保持恰好恒定。这增加在相同反应器长度下可提供的反应时间。
EP-A-2 196 455公开了在包含相对于流动方向基本旋转对称的反应空间的反应器中在高于胺的沸点下使光气和伯芳胺反应,其中在氨基转化成异氰酸酯基团的转化率为4%至80%的反应空间的区段中该反应混合物沿基本旋转对称的反应空间的轴的横截面平均流速为最大8米/秒,且其中在反应空间的这一区段中该反应混合物沿基本旋转对称的反应空间的轴的横截面平均流速始终低于在这一区段的起点处的横截面平均流速。
EP-A-1 935 876公开了通过使相应的伯胺与光气反应制备异氰酸酯的气相法,其中在高于胺的沸点下在0.05至15秒的平均接触时间内使光气和伯胺反应,其中所述反应绝热进行。
EP-A-2 408 738公开了如何可以避免光气由于含光气的料流在高温下的过长停留时间而离解成氯气和一氧化碳。通过将光气在大于300℃的温度下的停留时间减至最多5秒和通过将与光气接触的传热面的温度限制为高于要建立的光气温度最多20 K,应避免这一点。
EP-B-1 935 875公开了通过使相应的伯胺与光气在气相中反应制备异氰酸酯的方法,其中通过将反应混合物经过喷入有液体的冷却段导出反应空间,停止该反应,其中该冷却段中的直接冷却在串联的两个或更多个冷却区中一步实现(所谓的反应混合物的“骤冷”)。
WO 2013/029918描述了通过使相应的胺与光气反应制备异氰酸酯的方法,通过提高光气与胺的比率或将一种或多种惰性物质添加到光气和/或胺料流中,该方法也可毫无问题地在该气相装置的不同负荷下进行,特别是甚至在部分负荷范围内运行该装置时,该混合和/或反应也应在每种情况下优化的停留时间窗口内进行。此发明的方法应能以恒定的产品和工艺品质运行在不同负荷下的现有装置。这应免去提供多个具有不同标称容量的装置。
该申请教导了为在标称容量下运行生产装置而优化光气化的基本参数,特别例如反应物在各个装置中的停留时间,这在低于标称容量下运行该装置时会造成产率和产品一致性方面的问题(参见第2页第20至36行)。为了甚至在部分负荷(即与在标称容量下的运行相比减小的胺料流)下也能达到优化的 - 窄 - 停留时间窗口,建议增加光气料流和/或惰性成分(参见第3页第5至19行),确切来说,优选使得所有组分的总流量基本相当于在标称容量下的(参见第6页第4至8行)。该申请在第2页中提出的发明背景描述中虽然提到停机程序,但完全没有公开关于如何最有利地停止处于运行中的生产装置(即胺料流和光气料流等于0)的具体操作的技术教导。该申请中公开的技术措施(即增加光气料流和/或惰性成分)仅就生产装置在低于标称容量下运行的问题和如何有利地将在标称容量下运行的装置切换成在低于标称容量下运行(即胺和光气料流明显大于0)的问题进行考虑(参见实施例)。
尽管所述的现有技术方法成功地在不损失最终产物品质的情况下进行光气化,但除了少数例外,所描述的方法仅是处于正常运行下的方法。没有描述停机程序,即以稍后能顺利重启的方式安全地停止气相光气化装置。
本领域技术人员知道连续运行的工业过程不应该在非紧急情况下突然停止。气相光气化装置的停止(也称为停机)是工业日常中经常发生的程序,其不一定与打开或另一机械介入光气化装置的方式相关联。在实践中,停机的特征在于,与在生产装置的目标标称容量下的连续运行相比,光气相对于胺的过量可能偏差。特别在例如压力波动造成回混时出现这样的偏差。当要转化的胺的当前流量与在该装置的目标标称容量下要转化的胺的目标流量相比极小时,尤其观察到这一点。光气与胺的比率的这些量值波动不利,因为可沉淀出固体,如聚脲或胺盐酸盐。此外,在不恰当停止的情况下,会不合意地形成胺微滴。气相光气化装置的停止因此是关键工艺步骤,因为此处的错误会严重干扰稍后的重启和实际连续生产(例如由于为了确保反应物和产物经过该装置的足够流量所需的压力差的提高)。
因此,尽管在气相光气化领域中有各种进展,但仍需要进一步改进。考虑到这一需要,本发明提供一种运行用于使胺(2)与相对于胺(2)的伯氨基化学计算过量的光气(1)反应以产生相应的异氰酸酯(4)的气相光气化装置(100)的方法,其中所述气相光气化装置(100)至少包含
(i) 用于提供任选除光气(1)外还包含惰性物质(3)的气态光气料流(10)的装置1000,
(ii) 用于提供任选除胺(2)外还包含惰性物质(3)的气态胺料流(20)的装置2000,
(iii) 用于混合料流10和20的混合区(3100),其中所述混合区通过设备(1100、2100)分别与装置1000和装置2000相连,
(iv) 布置在混合区(3100)下游的用于之前混合的料流10和20的进一步反应的反应区(3200),
(v) 布置在反应区(3200)下游的用于终止所述反应的反应停止区(4000),
和任选的
(vi) 后处理部分(5000),其包含用于回收和再循环未转化的光气(1'')的设备(5100)和用于获得以纯净形式制成的异氰酸酯的设备(5200),
其中,在气相光气化装置(100)的正常运行中,光气气体料流(10)中的光气(1)优选是新鲜光气(1')和设备(5100)回收的再循环光气(1'')的混合物,
其中
通过运行下列步骤停止气相光气化装置(100):
(I) 将胺质量流量M'(2)降至0,同时使气态惰性气体料流(30)维持经过设备2100、混合区(3100)、反应区(3200)和反应停止区(4000);
(II) 在从M'(2)为0的时间点起计算经过相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的至少10倍,优选所述停留时间的至少30倍,更优选所述停留时间的至少60倍,最优选所述停留时间的至少90倍的时期后,才将离开装置1000的光气的质量流量M'(1)降至0,同时使气态惰性气体料流(30)维持经过设备1100、混合区(3100)、反应区(3200)和反应停止区(4000);
(III) 从离开装置1000的光气的质量流量M'(1)为0的时间点起计算,使至少来自(I)的惰性气体料流(30),优选来自(I)和(II)的惰性气体料流(30)维持至少相当于惰性气体料流(30)从进入混合区(3100)到离开反应停止区(4000)的停留时间的3倍,优选至少100倍,更优选至少200倍,最优选至少350倍的时期。
气相光气化根据本发明被理解为是指使胺光气化成相应的异氰酸酯的一种工艺方案,其中胺在气态下反应产生异氰酸酯并在该反应过程中,存在的所有组分(反应物、产物、中间产物、任选的副产物、任选的惰性物质)在经过反应区的过程中至少95.0质量%,优选至少98.0质量%,更优选至少99.0质量%,再更优选至少99.8质量%,特别是至少99.9质量%保持在气相中,在每种情况下基于存在的所有组分的总质量计。
组分在装置部分中的停留时间是指该组分在标准状态下的体积流量(表示为Vn/h,其中Vn代表在标准条件下的m3(“标准立方米”))和所涉装置部分的流经的内部体积(以m3表示)的商。在此,标准立方米Vn是在
• 1.01325巴的压力pn,
• 0%的空气湿度(干气体)和
• 273.15 K的温度Tn(tn = 0℃)(根据DIN 1343的标准条件,STPD)下的体积V为1立方米的气体量。
步骤(II)中相关的在气相光气化装置(100)的正常运行中的光气(1)的停留时间是由在正常运行中离开装置1000的光气(不存在可能的惰性物质成分)的已知体积流量和流经的装置的已知内部体积(包括连接它们的管道)确定的计算参数。以类似方式,通过计算确定步骤(III)中相关的惰性气体料流(30)的停留时间。这基于离开装置1000的质量流量M' (1)达到0时存在的惰性气体料流(30)的体积流量。在最简单的实施方案中,这一惰性气体料流(30)由来自(I)的惰性气体料流构成。但是,优选地,在(III)中,仍维持来自(II)的惰性气体料流。在这后一实施方案中,(III)中的惰性气体料流因此由来自(I)和(II)的合并的惰性气体料流构成。
合适的胺(2)尤其是异佛尔酮二胺、六亚甲基二胺、双(对氨基环己基)甲烷、甲苯二胺和二苯基甲烷二胺。
在本发明中,术语“气相光气化装置的停止”包括例如为了进行维护操作而停止处于运行中的气相光气化装置(其以特定时间点时的所需目标产量运行,表示为要转化的胺的目标质量流量M'目标(2) [例如t(胺)/h])所需的所有工艺步骤。气相生产装置(100)在M'目标(2)下的运行在本发明中被称作正常运行。M'目标(2)可以,但不需要,相当于在气相生产装置(100)的标称容量M'标称(2)下的M'目标(2)的值。生产装置的标称容量在专业领域中以每年要生产的产品吨数(“每年吨数”)表示,其中考虑所有可计划的装置停机。
本发明中关于可数参数的词语“一”仅被理解为不定冠词并且只有在明确说明时才是数词,例如通过加上“刚好一个”。例如,短语“一反应区”不排除存在多个(串联或并联)反应区的可能性。
对本发明而言重要的是,为了停止(“停机”)气相光气化装置(100),首先仅将胺质量流量M' (2)从数值M'目标(2)降至0。在停机时,始终在光气前停止胺,这防止胺回混到光气供应设备中并甚至在气相光气化装置(100)的停止过程中也确保光气相对于胺过量。通过根据本发明用惰性气体料流(30)充分冲洗胺供应设备,防止光气回流到胺供应设备中(回混)。从离开装置1000的光气的质量流量M' (1)为0的时间点起计算,最早在相当于来自(I)的惰性气体料流(30)在来自(I)和(II)的合并的惰性气体料流(30)的优选实施方案中从混合区(3100)的起点到离开反应停止区(4000)的停留时间的3倍,优选100 倍,更优选200倍,最优选350倍的时期后,停止用惰性气体料流(30)充分冲洗设备2100(优选还有设备1100)、混合区(3100)、反应区(3200)和反应停止区(4000)。
下面详细阐述本发明的步骤。各种实施方案在此可以任意互相组合,除非本领域技术人员从上下文中看出相反的意思。
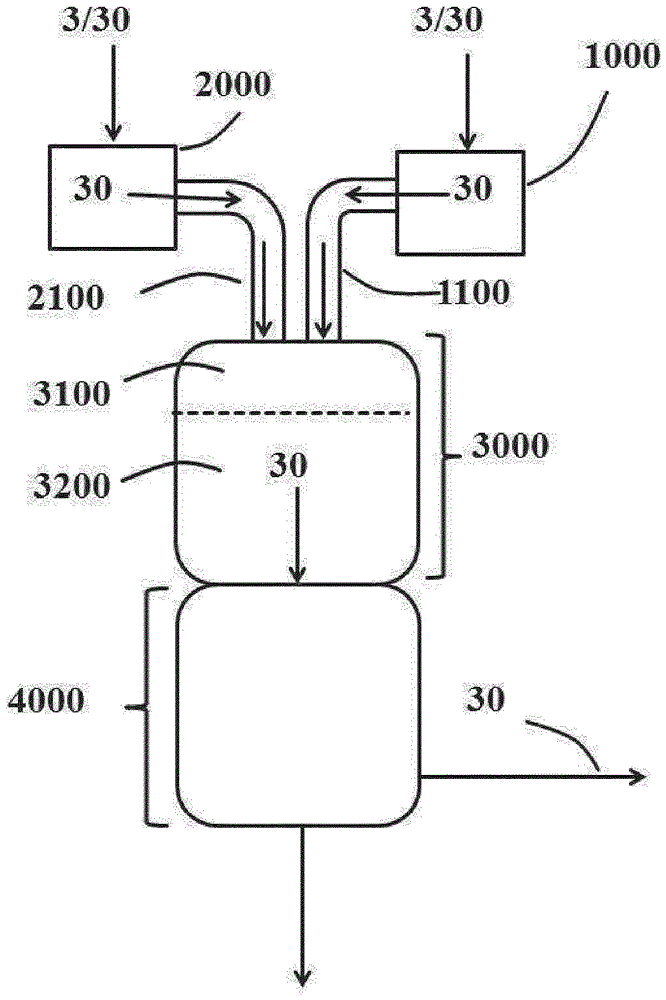
根据本发明,气相光气化装置(100)至少包含上文在(i)至(v)下列举的设备(也参见图1,其显示要根据本发明使用的气相光气化装置(100)的设备,包括优选存在的后处理部分和在正常运行中的物质料流)。
(i) 作为用于提供气态光气料流的装置(1000),原则上可以使用现有技术中已知并适用于将光气转化成气相的各种装置。优选通过如DE102009032413A1段落[0081]至[0118]中所述在蒸馏塔中的蒸馏或部分蒸发生成光气气体。可以由各种可想到的蒸发器,例如自然循环蒸发器、升膜蒸发器和降膜蒸发器实现在塔底的能量供应。降膜蒸发器尤其优选。
(ii) 作为用于提供气态胺料流的装置(2000),原则上可以使用现有技术中已知并适用于将胺转化成气相的各种装置,如本领域技术人员已知的蒸发装置。在一个优选实施方案中,装置2000包含蒸发设备和用于随后使胺(2)过热的设备。非常特别优选的是多级蒸发和过热系统,其中在蒸发和过热系统之间安装微滴分离器和/或该蒸发装置也具有微滴分离器的功能。合适的微滴分离器描述在例如"Droplet Separation", A. Bürkholz, VCH Verlagsgesellschaft, Weinheim – New York – Basle – Cambridge, 1989中。在离开流动方向上的最后一个过热器后,将预热至其目标温度的气态反应物料流(20)供入反应空间。
(iii) 可根据本发明使用的混合区(3100)可以以本领域技术人员已知的方式,优选如EP-A-2 196 455,尤其是段落[0047]至[0049]中,和EP-A-1 935 876,尤其是段落[0027]至[0029]中所述那样构造。混合区在正常运行中料流(10)和(20)初次彼此会合之处开始。在此位置,也称作混合区(3100)的入口,在停止程度的过程中,在引入步骤(II)后,混合来自(I)和(II)的惰性气体料流以产生共同的惰性气体料流,其在离开反应停止区(4000)前的停留时间构成计算步骤(III)的最小持续时间的基础。
(iv) 在混合区(3100)中初次彼此会合的胺和光气气体料流在停留时间装置(Verweilzeitapparat),即反应区(3200)中进一步反应。混合区(3100)和反应区(3200)也可优选如EP 2 196 455 A1,尤其是段落[0042]至[0049]中所述那样合并在单一装置,即反应器(3000)中。
根据本发明,将用于提供气态光气气体料流(1000)和胺气体料流(2000)的装置与混合区(3100)的设备1100和2100是适用于将来自装置1000和2000的气体料流(10)和(20)转移到混合区(3100)中的设备。这些设备除用于输送气体料流的管线外还优选包含确保光气气体料流(10)和胺气体料流(20)在混合区(3100)中的强力充分混合的喷嘴装置。可以将气体料流(10)和(20)的每个各自喷入混合区(3100)。但是,优选的是设备1100和2100的管线通往共用的喷嘴装置的实施方案(未显示在图1中)。在这一实施方案中,这两个气体料流之一,优选胺气体料流(20)经由布置在优选圆柱形容器的中心的内部喷嘴供入混合区(3100)。另一气体料流,优选光气气体料流(10)经过由内部喷嘴的外壁与该容器的内壁形成的环形空隙引入。这两个气体料流在内部喷嘴的出口孔处(= 混合区的起点)混合。这一实施方案例如显示在EP-A-1 449 826的图1中。在这种情况下,设备1100和2100部分互相集成并集成到混合区(3100)中。也可以如EP-A-1 449 826的图2中所示,使用由几个独立喷嘴构成的装置代替单一的中心喷嘴。例如在EP-A-2 196 455,尤其是段落[0047]至[0048]中,和EP-A-1 935 876,尤其是段落[0027]和[0028]中描述了可根据本发明用于设备1100和2100的进一步实施方案。
(v) 可根据本发明使用的反应停止区(4000)是本领域技术人员已知的。优选的是如EP 1 935 875 B1,尤其是段落[0024]和[0025]中所述的实施方案。在反应停止区(4000)中,将除异氰酸酯(4)外基本还包含氯化氢副产物(Koppelprodukt)和未转化的光气的反应粗产物(40)迅速冷却,优选通过将惰性溶剂(优选邻二氯苯,ODB)任选与一部分之前形成并再循环的异氰酸酯(4)一起喷入气体料流(40)。优选地,粗反应产物(40)在反应停止区(4000)中分离成气态组分(蒸气,50)和液体组分(60)。优选地,该反应停止区最早在离开装置1000的光气的质量流量M' (1)为0时停止运行。更优选地,该反应停止区在仅从离开装置1000的光气的质量流量M'(1)为0的时间点起计算相当于惰性气体料流(30)从进入混合区(3100)到离开反应停止区(4000)的停留时间的至少3倍,优选至少100倍,更优选至少200倍,最优选至少350倍的时期后。
在本发明的方法的一个特别优选的实施方案中,在反应停止区(4000)中获得的粗产物在相同的气相光气化装置(100)中后处理以从液体混合物(60)中分离异氰酸酯(4)。在这种情况下,气相光气化装置(100)另外包含
(vi) 后处理部分(5000)。
适用于后处理的装置描述在WO 2011/003532,尤其是第5页第19行至第28页第5行,以及EP 1 371 636 B1、EP 1 371 635 B1和EP 1 413 571 B1中,在每种情况下为全文。后处理部分(5000)可分成用于回收和再循环未转化的光气(和用于分离氯化氢副产物)的设备(5100)和用于获得以纯净形式制成的异氰酸酯(和任选用于再循环惰性溶剂)的设备(5200)。在图1中仅示意性标示后处理部分而没有下文给出的细节。特别地,后处理部分(5000)在设备5100中包含用于通过用惰性溶剂洗涤而从来自反应停止区(4000)的蒸气(50)中除去异氰酸酯的洗涤塔(5110)、用于通过吸收在惰性溶剂中(这导致氯化氢和惰性物(70)与光气分离)而从洗涤塔(5110)的蒸气中回收光气的光气吸收塔(5120)、用于分离光气和惰性溶剂的光气解吸塔(5130),以及在设备5200中尤其用于从粗制异氰酸酯中分离低沸物(尤其是来自反应停止区的惰性溶剂)的溶剂塔(5210)、尤其用于从在溶剂塔中预提纯的异氰酸酯中分离高沸物(例如含聚脲的残留物)以获得纯化的最终产物的精细提纯塔(5220)。
可以(未显示在图1中)将用于提供气态光气料流(10)的装置(1000)集成到光气解吸塔(5130)中以使来自后处理的含溶剂的光气料流与新鲜光气(1')一起蒸发并在光气解吸塔(5130)中蒸馏。所得气态光气经设备1100供入混合区,同时优选将分离出的惰性溶剂导入洗涤塔(5110)和/或光气吸收塔(5120)。
在以M'目标(2)的要转化的胺的所需目标质量流量运行气相光气化装置(100)的过程中,料流10和20的组成和质量流量优选互相匹配以在料流10中使光气(1)相对于料流20中的胺(2)的伯氨基以理论值的至少150%,更优选以理论值的160%至350%,最优选以理论值的170%至300%的化学计算过量存在。理论上,1摩尔光气与1摩尔氨基反应,意味着2摩尔光气理论上与1摩尔二胺反应。所需目标质量流量M'目标(2)可相当于基于常见装置利用率的情况下该气相光气化装置的标称容量,即在构造该装置所用于的特定时期内生产的异氰酸酯量(通常以每年吨数表示)。如果不利的销售状况所要求,所需目标质量流量M'目标(2)当然也可较低。这对本发明的方法不是基本的。例如,完全可设想,将本发明的方法应用于由于困难的销售环境而暂时仅以标称容量的50%运行的气相生产装置(100)。在该困难销售状况的持续期间,正常的生产运行随之相当于胺质量流量M'目标(2) = 0.5 · M'标称(2)。
气态光气料流(10)除光气(1)外还可含有惰性物质(3)。可根据本发明使用的惰性物质(3)除在室温和标准压力下已为气态的物质,如氮气、氦气或氩气外还包括在室温和标准压力下为液体的惰性有机溶剂的蒸气,例如任选具有卤素取代的芳烃,例如氯苯或二氯苯(所有异构体,优选邻二氯苯)。特别优选使用氮气稀释光气。可以以现有技术中常规的方式选择惰性物质(3)在光气气体料流(10)中的含量。
气态胺料流(20)除胺(2)外同样还可含有惰性物质(3)。在这种情况下,与上文对光气气体料流(10)所述相同的惰性物质(3)可行。如果这两个料流(10和20)都含有惰性物质(3),优选使用相同的惰性物质(3)。
气相光气化装置(100)在目标质量流量M'目标(2)的所需值下的运行可通过现有技术中已知的方法进行。优选地,为此,在避免回混下,将混合区(3100)中产生的反应混合物连续导过反应区(3200),并在其内优选在200℃至600℃的温度和80毫巴至2500毫巴的绝对压力下在0.05至15秒的平均接触时间内以绝热或等温方式,优选以绝热方式转化成包含所需异氰酸酯(4)的气态工艺产物。在EP 2 196 455 B1和EP 1 935 876 B1中描述了合适的实施方案。
在反应停止区(4000)中,将离开反应区(3200)的气态工艺产物(40)迅速冷却。这优选通过与温度保持在低于异氰酸酯(4)的沸点和高于相当于转化的胺的氨基甲酰氯的分解温度的惰性溶剂接触而实现。在EP 1 935 875 B1中描述了合适的实施方案。在这一步骤中,任选未冷凝的异氰酸酯(4)优选通过用洗涤液洗涤而从留在反应停止区中的气体混合物中分离并优选与在反应停止区(4000)中获得的冷凝物(60)合并。在EP 1 935 875 B1,尤其是段落[0024]和[0025]中描述了合适的实施方案。
此后,通过蒸馏从由此获得的粗制液体工艺产物(60)中分离所需异氰酸酯(4)。合适的实施方案是本领域技术人员已知的并例如描述在WO 2013/139703、EP 1 413 571 B1、EP 1 371 635 B1、EP 1 371 636 B1中。
在该反应中未转化的光气的大部分量留在反应停止区(4000)中获得的气相(50)中。优选的是,在优选存在的后处理部分5000的设备5100中后处理这种气相,以能将这种光气再循环。这一步骤是现有技术中充分已知的。已进一步在上文描述了合适的实施方案。
为了停止以这种方式运行的气相光气化装置,如今根据本发明运行下列步骤:
在本发明的方法的步骤(I)中,关闭进入混合和反应区的胺供应。这通过将引入装置2000的胺质量流量M'(2)从数值M'目标(2)降至0实现。连续或分段,优选连续进行这种降低。
在这一步骤(I)的过程中,使惰性气体料流(30)维持经过设备2100、混合区(3100)、反应区(3200)和反应停止区(4000)(也参见图2)。惰性气体料流(30)由至少一种惰性物质(3)构成,其中合适的惰性物质与如进一步在上文描述用于任选稀释气体料流(10)中的光气的那些相同。优选地,这一惰性气体料流(30)具有高于胺(2)在现有压力条件下的露点的温度。但是,优选地,该温度不会高到使仍存在于该装置中的反应物和产物具有热分解的危险。根据本发明优选的是200℃至600℃,更优选200℃至500℃,最优选250℃至500℃的温度。
可以通过(a)将优选在室温和标准压力下为液体的惰性物质(3)在低于步骤(I)中所需的惰性气体料流(30)的温度(即优选低于200℃)的温度T3下引入装置2000、在其中加热并然后将其导入设备2100和要惰性化的进一步装置部分来实现步骤(I)中的惰性气体料流(30)的维持。在变体(a)中,由此在装置2000中提供惰性气体料流(30)。当在正常运行中的反应过程中用在室温和标准压力下为液体的惰性物质(3)的蒸气稀释该反应混合物时,这一实施方案尤其有利。可以将惰性物质(3)以液体形式引入装置2000中并在此才将其蒸发。在变体(a)中特别合适的惰性物质(3)是惰性溶剂,如任选具有卤素取代的芳烃,例如氯苯或二氯苯(所有异构体,优选邻二氯苯)。在装置2000中,加热惰性物质(3)(即在以液体形式引入的情况下蒸发)以获得具有优选200℃至600℃,更优选200℃至500℃,最优选250℃至500℃的温度T30和优选100毫巴至3000巴,更优选150毫巴至2800毫巴,最优选200毫巴至2500毫巴的绝对压力p30的惰性气体料流(30)。料流(3)和(30)在化学组成上没有不同,仅在温度和任选压力方面不同。在将引入装置2000的料流(3)加热以使其以气体形式离开装置2000后,其被称作料流(30)。
也可以(b)将优选200℃至600℃,更优选200℃至500℃,最优选250℃至500℃的温度T30和优选100毫巴至3000毫巴,更优选150毫巴至2800毫巴,最优选200毫巴至2500毫巴的绝对压力p30的惰性气体料流(30)直接引入装置2000并从此处转移到设备2100和要惰性化的其它装置部分中。在这一实施方案中,也可以在上文规定的温度范围内进一步加热在装置2000中引入的惰性气体料流(30)。或者,也可以将惰性气体料流(30)直接供入设备2100。在后一实施方案中,优选在将胺M'(2)的质量流量降至0后,关闭装置2000和混合区(3100)之间的管线(其是设备2100的组成部分),尤其优选在装置2000后立即关闭。变体(a),无论配置如何,在正常运行中的反应过程中用在室温和标准压力下已为气态的惰性物质,如氮气、氦气或氩气稀释反应混合物时尤其有利。
直至来自装置2000的胺(2)的供应完全中断,才优选通过供入惰性物质料流(30)将装置2000中的压力p2000调节至高于混合区(3100)中的压力p3100的值。在这种情况下,p3100的值优选为80毫巴(绝对)至2500毫巴(绝对),而值p2000优选为100毫巴(绝对)至3000毫巴(绝对)。
在本发明的方法的步骤(II)中,将离开装置1000的光气的质量流量M'(1)从数值M'目标(1)降至0。这连续或分段,优选连续实现。在从M'(2)为0的时间点起计算经过相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的至少10倍,优选所述停留时间的至少30倍,更优选所述停留时间的至少60倍,最优选所述停留时间的至少90倍的时期后才进行步骤(II)。在此,使气态惰性气体料流(30)维持经过设备1100、混合区(3100)、反应区(3200)和反应停止区(4000)(也参见图3)。惰性气体料流(30)由至少一种气态惰性物质(3)构成,其中合适的惰性物质与如上文进一步描述用于任选稀释气体料流(10)中的光气的那些相同。
可以通过(a)将优选在室温和标准压力下为液体的惰性物质(3)在低于步骤(II)中所需的惰性气体料流(30)的温度的温度T3下引入装置1000、在其中加热并然后导入设备1100和要惰性化的进一步装置部分来实现步骤(II)中的惰性气体料流(30)的维持。优选地,步骤(II)的惰性气体料流(30)的温度为200℃至600℃,更优选200℃至500℃,最优选250℃至500℃。当在正常运行中的反应过程中用在室温和标准压力下为液体的惰性物质(3)的蒸气稀释该反应混合物时,这一实施方案尤其有利。可以将惰性物质(3)以液体形式引入装置1000中并在此才将其蒸发。在变体(a)中特别合适的惰性物质(3)是惰性溶剂,如任选具有卤素取代的芳烃,例如氯苯或二氯苯(所有异构体,优选邻二氯苯)。在装置1000中,加热惰性物质(3)(即在以液体形式引入的情况下蒸发)以获得具有优选200℃至600℃,更优选200℃至500℃,最优选250℃至500℃的温度T30和优选100毫巴至3000巴,更优选150毫巴至2800毫巴,最优选200毫巴至2500毫巴的绝对压力p30的惰性气体料流(30)。料流(3)和(30)在化学组成上没有不同,仅在温度和任选压力方面不同。在将引入装置1000的料流(3)加热以使其以气体形式离开装置1000后,其被称作料流(30)。如果步骤(I)根据变体(a)进行,优选也根据变体(a)进行步骤(II)。
也可以(b)将优选200℃至600℃,更优选200℃至500℃,最优选250℃至500℃的温度T30和优选100毫巴至3000毫巴,更优选150毫巴至2800毫巴,最优选200毫巴至2500毫巴的绝对压力p30的惰性气体料流(30)直接引入装置1000。在这一实施方案中,也可以在上文规定的温度范围内进一步加热在装置1000中引入的惰性气体料流(30)。或者,也可以将惰性气体料流(30)直接供入设备1100。变体(b),无论配置如何,在正常运行中的反应过程中用在室温和标准压力下已为气态的惰性物质,如氮气、氦气或氩气稀释反应混合物时尤其有利。如果步骤(I)根据变体(b)进行,优选也根据变体(b)进行步骤(II)。
在从M'(2)为0的时间点起计算经过相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的至少10倍,优选所述停留时间的至少30倍,更优选所述停留时间的至少60倍,最优选所述停留时间的至少90倍的时期后才进行步骤(II)的作用在于,可以将残留量的未转化的胺(2)光气化。
在一个优选实施方案中,气相光气化装置(100)包含后处理部分(5000)。在这种情况下,在气相光气化装置(100)的正常运行中,特别优选使用新鲜光气(1')和在设备5100中回收的再循环光气(1'')的混合物作为光气气体料流(10)中的光气(1)。这一工艺方案另外实现本发明方法的步骤(II)的特别有利的配置,即在胺质量流量M'(2)降至0后,
(II.a) 首先中断新鲜光气(1')的供应,然后
(II.b) 在从M'(2)为0的时间点起计算经过相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的至少10倍,优选所述停留时间的至少30倍,更优选所述停留时间的至少60倍,最优选所述停留时间的至少90倍的时期后,也才中断再循环光气(1'')的供应,以使离开装置1000的光气的质量流量M' (1)为0,
其中如进一步在上文描述,在步骤(II.a)和(II.b)的过程中使气态惰性气体料流(30)维持经过装置1100、混合区(3100)、反应区(3200)和反应停止区(4000)和任选至少经过设备5100的一些部分。
无论气相光气化装置(100)是否包含后处理部分5000,步骤(I)和(II)优选在小于12小时内,优选在小于6小时内,更优选在小于3小时内,最优选在小于1小时内进行。优选地,在步骤(I)和(II)中,使用相同组成的惰性气体料流(30)。
从离开装置1000的光气的质量流量M'(1)为0的时间点起计算,使来自(I)的惰性气体料流(30) (III)维持至少相当于惰性气体料流(30)从进入混合区(3100)到离开反应停止区(4000)的停留时间的3倍,优选100倍,更优选200倍,最优选350倍的时期。在一个优选实施方案中,同时,也维持来自(II)的惰性气体料流,其中在这种情况下来自(I)和(II)的合并的惰性气体料流的停留时间至关重要。在步骤(III)后 – 在冷却后 – 任选可以打开装置部分以进行维护操作。
来自步骤(I)和(II)的惰性气体料流(30)在离开反应停止区(4000)后,或如果存在,优选在经过后处理设备5100的至少一些部分后,优选经废气排放口排出。在包含后处理部分(5000)的本发明方法的优选实施方案中,来自步骤(I)和(II)的惰性气体料流(30)优选经过进一步在上文定义的装置洗涤塔(5110)和光气吸收塔(5120)并随后排出。
如果两个或更多个反应区(3200)并联运行,优选但不是必须地如上所述相继停止它们。辅助系统(如HCl吸收、光气吸收、任选的溶剂后处理或废气处理)的尺寸必须使得停机所需的惰性气体料流(30)的量可操作。
如果同时将所有反应物路径停机,会发生上述问题。光气可流入设备2100(优选胺喷嘴)并可造成堵塞、被聚脲附着等。此外,至少短暂地明显低于在连续生产中的目标质量流量M'目标(2)下所需的光气过量,以致由于干扰流量平衡和出现不受控的混合而产生副产物。在同时关闭这两个反应物料流时,扰乱反应物在反应空间中的停留时间。
在气相光气化装置(100)的停止中的本发明的程序因此为重启带来下列优点:
i) 避免设备2100(优选胺喷嘴)和混合区(3100)中的堵塞和因此避免由于必须停止该装置以清洁设备2100(优选胺喷嘴)而可能需要的多次启动。
ii) 由于(i),节省能源。
iii) 由于不需要由于附着物和沉积物的存在而反复停机和重启,提高气相光气化装置的生产率。
iv) 光气生产中的生产率提高和原料损失最小化。
v) 由于通过减少启动程序而降低光气化装置(100)上的热应力,使生产均质化且装置可靠性提高。
vi) 降低的副产物形成和对产物的热应力缩短,伴随着相对收率的提高。
vii) 避免或减少该设备中(例如装置2000和设备2100中或反应停止前的反应区中)的沉淀物、附着物和堵塞,伴随着该方法的开工时间(Standzeit)的延长。
viii) 清洁该设备后的较少废物(例如要除去的聚脲较少,避免可能引发腐蚀的洗涤水)。
ix) 避免由于反复不良启动和停机而可能形成的不符合规格的产品。这样的劣质启动产品因此不必与优质多异氰酸酯掺混(verschnitten)或甚至在最糟的情况下焚化。
因此,本发明的方法通过避免在气相光气化装置的停止过程中光气(1)回混到胺气体料流(20)中而能顺利重启该装置并随后以技术上顺利的方式后处理形成的粗制异氰酸酯,其中停工时间降低或在理想的情况下无停工时间并直接具有高的最终产物品质。由此可避免或至少在范围上减少在停机过程中的复杂清洁操作。
实施例
以ppm或%计的含量数据是基于相关的各物质(料流)的总质量计的质量含量。
用于在正常运行中的"运行中(eingefahren)的"气相生产装置(100)制备甲苯二异氰酸酯(TDI)的一般条件
(也参见图1(简化图))
甲苯二胺(TDA)(2)在胺蒸发器(2000)中与氮气(3)一起连续蒸发。将含有12 t/h的气态TDA (2)的由此获得的胺气体料流(20)经由在其朝向光气化反应器(3000)的末端存在胺喷嘴的导管(2100)连续喷入光气化反应器(3000)。TDA料流(20)从离开蒸发器(2000)到离开胺喷嘴的停留时间为5秒。同时,经由如EP-A-1 362 847中公开的那样使用的光气精馏器(Gleichrichter),将61 t/h的气态光气料流(10)连续喷入光气化反应器(3000)。所用光气是新鲜光气(1')和在后处理部分(5000)中回收的光气(1'')的混合物。在这种情况下,良好混合这两种反应物,并且没有回混。气态TDA料流(20)在喷嘴口的温度为380℃(TDA在喷嘴口的进料中在此温度下的停留时间为大约1秒)。气态光气(10)在离开光气精馏器时具有320℃的温度,其中热光气在最后一个光气过热器和光气精馏器之间的停留时间为2秒。来自料流(10)和(20)的气态混合物在气相反应器(3000)中具有8秒的停留时间并在1692毫巴的绝对压力下反应产生气态反应混合物(40)。随后的反应停止区(4000)包含两段“骤冷”,其中通过喷入邻二氯苯(ODB)将气态反应混合物(40)冷却至168℃,以使其冷凝且粗制TDI和ODB的混合物(60)收集在底部容器(4100)中。过量光气、在该反应中形成的氯化氢和惰性物在这些条件下从底部容器(4100)中基本脱气,其中借助内部构件减少TDI的夹带。这一工艺残留气体料流(50)如WO 2011/003532,第11页第24至25行中所述那样后处理(5100)以回收夹带的TDI、光气和氯化氢。来自底部容器(4100)的混合物(60)如EP 1 413 571 B1中所述那样后处理(5200),从而以15.6 t/h的流量提供TDI (4)。
由此制备的TDI (4)通常具有> 99.97%的纯度(气相色谱法,GC)、< 5 ppm的ODB残留溶剂含量(GC)、< 10 ppm的可水解氯的残留氯含量(根据ASTM D4663滴定)、< 5 ppm的结合氯的酸度(根据ASTM D5629滴定)和作为Hazen值测得的< 15的色值(根据DIN EN ISO 6271测定)。
对比例1: 在胺供应前停止光气供应以停止气相光气化装置(100)
气相光气化装置(100)如用于制备TDI的一般条件中所述那样在15.6 t/h TDI的生产量下运行,然后如下停止:
中断光气进料,同时胺进料还维持大约另外2分钟。胺TDA在此期间不再完全光气化,且形成的中间产物和副产物堵塞混合区(3100)、反应区(3200)和随后的反应停止区(“骤冷”,4000)。从反应物TDA气体料流(20)和光气气体料流(10)进入光气化反应器(3000)的入口经过反应停止区(4000)底部的蒸气出口直到TDI洗涤塔(5110)的压差在中断光气气体料流(10)并继续胺气体料流(20)的短时间(大约2分钟)内升至913毫巴,而非标准运行中的10毫巴。在能够重启前必须进行反应器及其外围设备的清洁。
对比例2: 同时停止胺和光气供应以停止气相光气化装置(100)
气相装置(100)如用于制备TDI的一般条件中所述那样在15.6 t/h TDI的生产量下运行,然后如下停止:
关闭光气发生器。此后,同时中断向光气化反应器(3000)的胺和光气供应。随后,光气反应器(3000)用氮气惰性化直至光气化反应器及其管线外围已脱除反应物和产物。
在短暂的反应中断后,包括下游热交换器(2000)的胺蒸发器和包括胺喷嘴的设备2100在380℃的设定温度下进料氮气。光气化反应器(3000)无反应物和产物并用热氮气惰性化。启动胺蒸发器中的胺蒸发,以使TDA在300℃下蒸发。随后,TDA在进一步热交换器中逐步加热至380℃并作为1691毫巴(绝对)的压力的气态TDA料流(20)经胺喷嘴喷入光气化反应器(3000)。在45分钟内,引入光气化反应器(3000)中的TDA (2)的质量流量从0 t/h连续提高到12 t/h。在TDA气体料流(20)经胺喷嘴进入光气化反应器(3000)前不久,打开光气路径并将光气气体料流(10)以61 t/h的质量流量和以反应器入口处320℃的温度和1691毫巴(绝对)的压力喷入光气化反应器(3000)。该装置在45分钟后达到正常运行状态。在7天后,由于从反应物TDA气体料流(20)和光气气体料流(10)进入光气化反应器(3000)的入口经过反应停止区(4000)底部的蒸气出口直到TDI洗涤塔(5110)的压差随时间升至793毫巴而非在标准运行中的10毫巴,必须停止该装置,且蒸发光气和TDA并将气体料流(10)和(20)转移到光气化反应器(3000)中所需的能量几乎无法提高(随压力提高,TDA (2)的沸点限制蒸发器处理量)。在该装置停止和打开后,在胺喷嘴的出口处、沿反应器空间的表面和在骤冷器(Quenchen)表面上发现含聚脲的严重(massiv)沉积物。
对比例3: 在光气供应前停止胺供应以停止气相光气化装置(100),但不用惰性气体冲洗胺供应设备(光气回流到胺喷嘴中)
气相生产装置(100)如用于制备TDI的一般条件中所述那样在15.6 t/h TDI的生产量下运行,然后如下停止:
通过关闭在胺蒸发器(2000)后的反应物引导管线(设备2100的组成部分),停止向光气化反应器(3000)的胺供应。不进行设备2100的惰性气体冲洗。此后,停止光气反应器并因此中断新鲜光气(1')的供应。在15分钟后(相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的大约112倍),通过也中断向光气化反应器(3000)供应再循环光气(1'')而使光气循环停止。随后,光气化反应器(3000)用氮气惰性化。
在短暂停机后,通过使来自出自反应停止区(4000)的蒸气(50)在320℃下的后处理(5100)的再循环光气(1'')经过光气化反应器(3000)、反应停止区(4000)回到该后处理,建立光气的循环。在反应停止区(4000)中,在此过程中,只有在反应混合物(40)的流动方向上的第二骤冷在运转,因此将光气料流冷却。在这一光气循环中,使61 t/h的光气循环。在此过程中,用氮气流(30)冲洗包括胺喷嘴的导管2100。光气化反应器(3000)一旦调温到320℃,就通过将预热到220℃的液体TDA (2)与氮气一起送入胺蒸发器(2000)、在此处借助热交换器在300℃下蒸发并然后借助另一热交换器加热到410℃而开始胺蒸发。由此获得的TDA料流(20)在1654毫巴的绝对压力下经胺喷嘴喷入光气化反应器(3000)。在气相光气化装置的启动过程中(即直到胺质量流量达到M'目标(2),这在45分钟后发生)引入光气化反应器(3000)的TDA (2)的量从0 t/h连续增加到12 t/h,其中在启动结束时光气化反应器(3000)中的工作压力为1641毫巴(绝对)。在TDA气体料流(20)在启动过程中朝光气化反应器(3000)的方向初次离开胺喷嘴前不久,启动在反应混合物(40)的流动方向上的第一骤冷。通过新鲜光气(1')和在后处理中回收的光气(1'')的混合物替代在该装置启动后消耗的光气。在60分钟后,15.6 t/h的TDI (4)离开后处理阶段的最后一个蒸馏塔。
在启动后,胺蒸发(2000)和光气化反应器(3000)之间的压差始终进一步提高。在3小时后,由于胺喷嘴处的工作压力已升至2.5巴(绝对),必须停止光气化反应器(3000)。在该装置停止和打开后,在胺喷嘴的出口孔中和在通往胺喷嘴的管线中发现炭化的残留物。这可归因于在启动阶段中光气回流到设备2100中,以致形成堵塞胺喷嘴的出口孔的TDI沉积物。
对比例4: 在光气供应前停止胺供应的情况下(在维持胺供应设备的惰性气体冲洗的同时(防止光气回流到胺喷嘴中))的气相光气化装置(100)的启动,但光气的后反应时间(停留时间)太短
气相光气化装置(100)如用于制备TDI的一般条件中所述那样在15.6 t/h TDI的生产量下运行并如下停止:
通过关闭在胺蒸发器(2000)后的反应物引导管线(设备2100的组成部分),中断向光气化反应器(3000)的胺供应。在此过程中,使气态氮气料流维持经过设备2100、混合区(3100)、反应区(3200)、反应停止区(4000)和设备5100的一些部分(洗涤塔5120和光气吸收塔5120,都未显示在图1中)(本发明方法的步骤(I))。在胺质量流量M'(2)已降至0后,停止光气发生器并因此中断新鲜光气(1')的供应(本发明方法的步骤(II.a))。在30秒后(相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的大约4倍),通过也中断向光气化反应器(3000)供应再循环光气(1'')而使光气循环停止(本发明方法的步骤(II.b))。在整个时间过程中,设备1100、反应器(3000)、反应停止区(4000)和设备5100的一些部分(洗涤塔5120和光气吸收塔5120,都未显示在图1中)用氮气充分冲洗,其通过从光气吸收塔(5120)中排出而离开设备5100。在已中断光气循环后,使来自步骤(I)、(II.a)和(II.b)的氮气料流维持另外1小时(相当于合并的惰性气体料流(30)从进入混合区(3100)到离开反应停止区(4000)的停留时间的大约400倍)(本发明方法的步骤(III)),此后光气化反应器(3000)已脱除反应物和产物。
在该装置的另一部分中的1天停机维护后,如实施例3中所述那样再次启动气相光气化装置。
在启动结束时,即在开启结束时,光气化反应器(3000)中的工作压力为1651毫巴(绝对)。在13天后,由于从反应物TDA气体料流(20)和光气气体料流(10)进入光气化反应器(3000)的入口经过反应停止区(4000)底部的蒸气出口直到TDI洗涤塔(5110)的压差随时间升至803毫巴而非在标准运行中的10毫巴,必须停止该装置,且蒸发光气和TDA并将气体料流(10)和(20)转移到光气化反应器(3000)中所需的能量几乎无法提高(随压力提高,TDA (2)的沸点限制蒸发器处理量)。在该装置停止和打开后,在胺喷嘴的出口处、沿反应器空间的表面和在骤冷器表面上发现含聚脲的严重沉积物。
实施例5(本发明)
气相光气化装置(100)如用于制备TDI的一般条件中所述那样在15.6 t/h TDI的生产量下运行并如下停止:
通过关闭在胺蒸发器(2000)后的反应物引导管线(设备2100的组成部分),中断向光气化反应器(3000)的胺供应。在此过程中,使气态氮气料流维持经过设备2100、混合区(3100)、反应区(3200)、反应停止区(4000)和设备5100的一些部分(洗涤塔5120和光气吸收塔5120,都未显示在图1中)(本发明方法的步骤(I))。在胺质量流量M'(2)已降至0后,停止光气发生器并因此中断新鲜光气(1')的供应(本发明方法的步骤(II.a))。在15分钟后(相当于在气相光气化装置(100)的正常运行中光气(1)从离开装置1000到离开反应停止区(4000)的停留时间的大约112倍),通过也中断向光气化反应器(3000)供应再循环光气(1'')而使光气循环停止(本发明方法的步骤(II.b))。在整个时间过程中,设备1100、反应器(3000)、反应停止区(4000)和设备5100的一些部分(洗涤塔5120和光气吸收塔5120,都未显示在图1中)用氮气充分冲洗,其通过从光气吸收塔(5120)中排出而离开设备5100。在已中断光气循环后,使来自步骤(I)、(II.a)和(II.b)的氮气料流维持另外1小时(相当于合并的惰性气体料流(30)从进入混合区(3100)到离开反应停止区(4000)的停留时间的大约400倍)(本发明方法的步骤(III)),此后光气化反应器(3000)已脱除反应物和产物。
在该装置的另一部分中的1天停机维护后,如实施例3中所述那样再次启动气相光气化装置。
在实施例5中,在停止过程中没有观察到胺喷嘴的堵塞和沉积物在光气化反应器中的形成。该装置可以无问题地重启并运行数月的另一光气化周期。明显减少不想要的副产物,如聚脲等的形成并且启动产品具有高品质,以致不必须与稍后生产的TDI掺混。
- 还没有人留言评论。精彩留言会获得点赞!