核基因输出的定向增加的制作方法
交叉引用
本申请要求2014年10月3日提交的第62/059,847号美国临时申请的权益,该申请通过引用并入本文。
背景技术:
一些遗传病由单倍性不足而引起,其中仅存在基因的一个功能性拷贝,并且该单个拷贝无法产生足够的基因产物。例如,这可由半合子缺失而引起,其中该基因的一个拷贝丢失。其他遗传病则由改变基因产物使其仅具有部分功能的突变而引起。
技术实现要素:
如本文所述,可使用反义寡聚体(aso),通过促进在含内含子的基因的内含子剪接位点处的组成性剪接(采用野生型序列)以增加基因产物的表达,来增加蛋白质或功能rna(在非蛋白质编码基因的情况下)的产生量。针对用于这些方法而描述的aso促进组成性剪接且不校正由突变导致的异常剪接,或促进组成性剪接且不调节其它剪接。因此,本文所述的方法可用来治疗由基因产物的表达减少或活性不足所导致的病况。
本文描述了增加在细胞中表达由前mrna编码的靶蛋白的方法,该前mrna包含至少一个保留内含子(ric前mrna);保留内含子是当一个或多个其他内含子被剪接掉(去除)时仍然存在的内含子。靶蛋白的表达依赖于细胞核的前mrna中全部内含子的完全剪接(去除)以生成成熟mrna,成熟mrna随后输出到细胞质并翻译为靶蛋白。内含子的未充分剪接(去除)导致含保留内含子(ric)的前mrna主要蓄积在细胞核中,并且如果被输出到细胞质则其降解,使得ric前mrna未翻译为靶蛋白。用本文方法所述的反义寡聚体(aso)处理可以促进将保留内含子从前mrna转录物(包含一个或多个内含子的前mrna种类)上剪接,以及导致mrna的增加,该增加的mrna得到翻译以提供更高水平的靶蛋白。
在实施方案中,所述方法是增加具有含保留内含子的前mrna(ric前mrna)的细胞表达靶蛋白或功能rna的方法,该ric前mrna包含保留内含子、位于保留内含子的5’剪接位点侧翼的外显子、位于保留内含子的3’剪接位点侧翼的外显子,并且其中该ric前mrna编码该靶蛋白或功能rna。在实施方案中,该方法包括使所述细胞与同编码靶蛋白或功能rna的ric前mrna的靶向部分互补的aso相接触,借此将保留内含子从编码靶蛋白或功能rna的ric前mrna中组成性剪接,从而增加编码靶蛋白或功能rna的mrna的水平,以及增加该细胞中靶蛋白或功能rna的表达。在实施方案中,所述细胞在受试者中或来自受试者,并且所述方法是治疗该受试者以增加该受试者的细胞表达靶蛋白或功能rna的方法。在实施方案中,所述细胞在具有由靶蛋白的量或活性缺陷或功能rna的量或活性缺陷引起的病况的受试者中,或来自该受试者。在实施方案中,该靶蛋白或该功能rna是补偿蛋白或补偿功能rna,其功能性地增加或代替在受试者中的量或活性缺陷的靶蛋白或功能rna。
在实施方案中,由靶蛋白的量或活性缺陷或功能rna的量或活性缺陷引起的病况并非由aso所靶向的保留内含子的其它剪接或异常剪接而引起的病况。在实施方案中,由靶蛋白的量或活性缺陷或功能rna的量或活性缺陷引起的病况并非由在编码该靶蛋白或功能rna的ric前mrna中的任何保留内含子的其它剪接或异常剪接而引起的病况。
在实施方案中,靶蛋白的量的缺陷由该靶蛋白的单倍性不足所导致,其中该受试者具有编码功能性靶蛋白的第一等位基因,以及不产生该靶蛋白的第二等位基因,或编码非功能性靶蛋白的第二等位基因,并且其中所述反义寡聚体与由第一等位基因所转录的ric前mrna的靶向部分结合。
在其他实施方案中,受试者具有由常染色体隐性病症引起的病况,该常染色体隐性病症由靶蛋白的量或功能缺陷导致,其中该受试者具有a)第一突变体等位基因,由其i)产生靶蛋白的水平与由野生型等位基因产生相比降低,ii)产生与对应的野生型蛋白相比功能降低的形式的靶蛋白,或iii)不产生靶蛋白,以及b)第二突变体等位基因,由其i)产生靶蛋白的水平与由野生型等位基因产生相比降低,ii)产生与对应的野生型蛋白相比功能降低的形式的靶蛋白,或iii)不产生靶蛋白,并且其中ric前mrna由该第一等位基因和/或该第二等位基因转录。在实施方案中,与对应的野生型蛋白相比,以降低的水平产生功能降低的形式的靶蛋白。
在实施方案中,与对应的野生型蛋白相比,产生功能降低的形式的靶蛋白。在其他实施方案中,与对应的野生型蛋白相比,产生完全功能性形式的靶蛋白。
在实施方案中,功能rna的量的缺陷由该功能rna的单倍性不足所导致,其中受试者具有编码功能性的功能rna的第一等位基因,以及不产生该功能rna的第二等位基因,或编码非功能性的功能rna的第二等位基因,并且其中所述反义寡聚体与由该第一等位基因所转录的ric前mrna的靶向部分结合。
在其他实施方案中,受试者具有由常染色体隐性病症引起的病况,该常染色体隐性病症由功能rna的量或功能缺陷导致,其中该受试者具有a)第一突变体等位基因,由其i)产生功能rna的水平与由野生型等位基因产生相比降低,ii)产生与对应的野生型蛋白相比功能降低的形式的功能rna,或iii)不产生功能rna,以及b)第二突变体等位基因,由其i)产生功能rna的水平与由野生型等位基因产生相比降低,ii)产生与对应的野生型蛋白相比功能降低的形式的功能rna,或iii)不产生功能rna,并且其中ric前mrna由该第一等位基因和/或该第二等位基因转录。在实施方案中,与对应的野生型功能rna相比,以降低的水平产生功能降低的形式的功能rna。
在实施方案中,产生与对应的野生型蛋白相比功能降低的形式的功能rna。在其他实施方案中,产生与对应的野生型蛋白相比为完全功能性形式的功能rna。
在实施方案中,ric前mrna的靶向部分在保留内含子中相对于该保留内含子的5’剪接位点的+6区域至相对于该保留内含子的3’剪接位点的-16区域之内。在实施方案中,ric前mrna的靶向部分在保留内含子中相对于该保留内含子的5’剪接位点的+6至+100区域内;或在相对于该保留内含子的3’剪接位点的-16至-100区域内。在实施方案中,ric前mrna的靶向部分在位于该保留内含子的5’剪接位点侧翼的外显子中的+2e至-4e区域内;或在位于该保留内含子的3’剪接位点侧翼的外显子中的+2e至-4e区域内。
在实施方案中,所述反义寡聚体并不是通过调节由编码功能rna或靶蛋白的基因转录的前mrna的其它剪接来增加该靶蛋白或该功能rna的量。在实施方案中,该反义寡聚体并不是通过调节由编码靶蛋白或功能rna的基因的突变所导致的异常剪接来增加该靶蛋白或该功能rna的量。
在实施方案中,所述ric前mrna由全长前mrna的部分剪接或野生型前mrna的部分剪接所产生。在实施方案中,编码靶蛋白或功能rna的mrna是全长成熟mrna或野生型成熟mrna。在实施方案中,所产生的靶蛋白是全长蛋白或野生型蛋白。在实施方案中,所产生的功能rna是全长功能rna或野生型功能rna。
在实施方案中,与编码在对照细胞中所产生的靶蛋白或功能rna的mrna的总量或成熟mrna的总量相比,编码在与所述反义寡聚体相接触的细胞中所产生的靶蛋白或功能rna的mrna的总量或成熟mrna的总量增加至约1.1至约10倍、约1.5至约10倍、约2至约10倍、约3至约10倍、约4至约10倍、约1.1至约5倍、约1.1至约6倍、约1.1至约7倍、约1.1至约8倍、约1.1至约9倍、约2至约5倍、约2至约6倍、约2至约7倍、约2至约8倍、约2至约9倍、约3至约6倍、约3至约7倍、约3至约8倍、约3至约9倍、约4至约7倍、约4至约8倍、约4至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。
在实施方案中,与编码在对照细胞中所产生的靶蛋白或功能rna的mrna的总量相比,编码在与所述反义寡聚体相接触的细胞中所产生的靶蛋白或功能rna的mrna的总量增加至约1.1至约10倍、约1.5至约10倍、约2至约10倍、约3至约10倍、约4至约10倍、约1.1至约5倍、约1.1至约6倍、约1.1至约7倍、约1.1至约8倍、约1.1至约9倍、约2至约5倍、约2至约6倍、约2至约7倍、约2至约8倍、约2至约9倍、约3至约6倍、约3至约7倍、约3至约8倍、约3至约9倍、约4至约7倍、约4至约8倍、约4至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。
在实施方案中,与编码在对照细胞中所产生的靶蛋白或功能rna的成熟mrna的总量相比,编码在与所述反义寡聚体相接触的细胞中所产生的靶蛋白或功能rna的成熟mrna的总量增加至约1.1至约10倍、约1.5至约10倍、约2至约10倍、约3至约10倍、约4至约10倍、约1.1至约5倍、约1.1至约6倍、约1.1至约7倍、约1.1至约8倍、约1.1至约9倍、约2至约5倍、约2至约6倍、约2至约7倍、约2至约8倍、约2至约9倍、约3至约6倍、约3至约7倍、约3至约8倍、约3至约9倍、约4至约7倍、约4至约8倍、约4至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。
在实施方案中,与对照细胞所产生的靶蛋白或功能rna的量相比,与所述反义寡聚体相接触的细胞所产生的靶蛋白或功能rna的总量增加至约1.1至约10倍、约1.5至约10倍、约2至约10倍、约3至约10倍、约4至约10倍、约1.1至约5倍、约1.1至约6倍、约1.1至约7倍、约1.1至约8倍、约1.1至约9倍、约2至约5倍、约2至约6倍、约2至约7倍、约2至约8倍、约2至约9倍、约3至约6倍、约3至约7倍、约3至约8倍、约3至约9倍、约4至约7倍、约4至约8倍、约4至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。
在实施方案中,所述方法包括使具有ric前mrna的细胞与包含主链修饰的反义寡聚体接触,该主链修饰包含硫代磷酸酯键或磷二酰胺键。在实施方案中,该反义寡聚体包含磷二酰胺吗啉基(pmo)、锁定核酸(lna)、肽核酸(pna)、2’-o-甲基、2’-氟或2’-o-甲氧乙基部分。在实施方案中,该反义寡聚体包含至少一个经修饰的糖部分。在相关的实施方案中,每一个糖部分均为经修饰的糖部分。
在实施方案中,所述反义寡聚体由8至50个核碱基组成。在实施方案中,该反义寡聚体由8至40个核碱基、8至35个核碱基、8至30个核碱基、8至25个核碱基、8至20个核碱基、8至15个核碱基、9至50个核碱基、9至40个核碱基、9至35个核碱基、9至30个核碱基、9至25个核碱基、9至20个核碱基、9至15个核碱基、10至50个核碱基、10至40个核碱基、10至35个核碱基、10至30个核碱基、10至25个核碱基、10至20个核碱基、10至15个核碱基、11至50个核碱基、11至40个核碱基、11至35个核碱基、11至30个核碱基、11至25个核碱基、11至20个核碱基、11至15个核碱基、12至50个核碱基、12至40个核碱基、12至35个核碱基、12至30个核碱基、12至25个核碱基、12至20个核碱基或12至15个核碱基组成。在实施方案中,该反义寡聚体与编码所述蛋白质的ric前mrna的靶向部分至少80%、至少85%、至少90%、至少95%、至少98%、至少99%或100%互补。
在任何前述方法中,所述细胞可包含由编码靶蛋白或功能rna的基因所转录的一群ric前mrna,其中该群ric前mrna包含两个或更多个保留内含子,并且其中所述反义寡聚体与该群ric前mrna中最丰富的保留内含子结合。在这些实施方案中,该反义寡聚体与最丰富的保留内含子的结合可诱导所述两个或更多个保留内含子从该群ric前mrna中剪接掉,以产生编码该靶蛋白或功能rna的mrna。
在其他实施方案中,所述细胞包含由编码靶蛋白或功能rna的基因所转录的一群ric前mrna,其中该群ric前mrna包含两个或更多个保留内含子,并且其中所述反义寡聚体与该群ric前mrna中第二丰富的保留内含子结合。在这些实施方案中,该反义寡聚体与第二丰富的保留内含子的结合可诱导所述两个或更多个保留内含子从该群ric前mrna中剪接掉,以产生编码该靶蛋白或功能rna的mrna。
在前述方法中,所述病况可以是疾病或病症。在这些实施方案中,该疾病或病症可选自:血栓性血小板减少性紫癜、复合型结节性硬化症、多囊肾病、家族性自主神经功能异常、10型视网膜色素变性、11型视网膜色素变性、囊性纤维化、视网膜母细胞瘤、家族性腺瘤性息肉病、蛋白s缺陷症、β地中海贫血和镰状细胞病。在相关的实施方案中,所述靶蛋白和ric前mrna由选自adamts13、tsc1、pkd1、ikbkap、impdh1、prpf31、cftr、rb1、apc、pros1、nedd4l、hbg1、hbg2和hbb的基因编码。在实施方案中,所述反义寡聚体可与选自seqidno:1-102的ric前mrna的一部分结合。
在实施方案中,任何前述方法进一步包括评估蛋白质表达。
在一些实施方案中,所述受试者是人。在其他实施方案中,该受试者是非人类动物。在实施方案中,所述反义寡聚体通过受试者的玻璃体内注射、鞘内注射、腹膜内注射、皮下注射或静脉内注射来施用。在实施方案中,所述细胞是离体的。
在实施方案中,在位于5’剪接位点侧翼的外显子的-3e至-1e和保留内含子的+1至+6处的9个核苷酸与相应的野生型序列相同。在实施方案中,在保留内含子的-15至-1和位于3’剪接位点侧翼的外显子的+1e处的16个核苷酸与相应的野生型序列相同。
本文描述了包含用于本文所述方法的反义寡聚体的组合物。还描述了包含该反义寡聚体和赋形剂的药物组合物。在实施方案中,包含该反义寡聚体的组合物意在用于增加细胞表达靶蛋白或功能rna的方法中,以治疗受试者中与缺陷蛋白质或缺陷功能rna相关的病况,其中该缺陷蛋白质或缺陷功能rna是该受试者中量或活性的缺陷,其中该反义寡聚体增强编码靶蛋白或功能rna的含保留内含子的前mrna(ric前mrna)的组成性剪接,其中该靶蛋白为:(a)所述缺陷蛋白质;或(b)在功能上增加或代替该受试者中的缺陷蛋白质的补偿蛋白;并且其中该功能rna为:(a)所述缺陷rna;或(b)在功能上增加或代替该受试者中的缺陷功能rna的补偿功能rna;其中该ric前mrna包含保留内含子,位于5’剪接位点侧翼的外显子,以及位于3’剪接位点侧翼的外显子,并且其中将该保留内含子从编码该靶蛋白或该功能rna的ric前mrna中剪接,从而增加该受试者中靶蛋白或功能rna的产生量或活性。
在实施方案中,包含所述反义寡聚体的组合物意在用于治疗受试者中与靶蛋白或功能rna相关的疾病或病症的方法,该方法包括增加该受试者的细胞表达该靶蛋白或功能rna的步骤,其中该细胞具有含保留内含子的前mrna(ric前mrna),该ric前mrna包含保留内含子,位于该保留内含子的5’剪接位点侧翼的外显子,位于该保留内含子的3’剪接位点侧翼的外显子,并且其中该ric前mrna编码该靶蛋白或功能rna,该方法包括使所述细胞与反义寡聚体相接触,借此将该保留内含子从编码该靶蛋白或功能rna的ric前mrna转录物中组成性剪接,从而增加该受试者的细胞中编码该靶蛋白或功能rna的mrna的水平,并且增加该受试者的细胞中该靶蛋白或功能rna的表达。
在实施方案中,所述包含反义寡聚体的组合物意在用于治疗受试者中由靶蛋白或功能rna的量或活性缺陷所导致的病况的方法。在实施方案中,该病况是疾病或病症。在实施方案中,该疾病或病症选自:血栓性血小板减少性紫癜、复合型结节性硬化症、多囊肾病、家族性自主神经功能异常、10型视网膜色素变性、11型视网膜色素变性、囊性纤维化、视网膜母细胞瘤、家族性腺瘤性息肉病、蛋白s缺陷症、β地中海贫血和镰状细胞病。在实施方案中,该组合物意在用于一种方法,其中靶蛋白和ric前mrna由选自adamts13、tsc1、pkd1、ikbkap、impdh1、prpf31、cftr、rb1、apc、pros1、nedd4l、hbg1、hbg2和hbb的基因编码。
在实施方案中,所述组合物的反义寡聚体靶向ric前mrna的一部分,该部分在保留内含子中相对于该保留内含子的5’剪接位点的+6区域至相对于该保留内含子的3’剪接位点的-16区域内。在实施方案中,该组合物的反义寡聚体靶向ric前mrna的一部分,该部分在保留内含子中相对于该保留内含子的5’剪接位点的+6至+100区域内;或在相对于该保留内含子的3’剪接位点的-16至-100区域内。在实施方案中,所述反义寡聚体靶向ric前mrna的一部分,该部分在所述至少一个保留内含子的5’剪接位点下游约100个核苷酸至所述至少一个保留内含子的3’剪接位点上游约100个核苷酸的区域内。在实施方案中,所述ric前mrna的靶向部分在位于该保留内含子的5’剪接位点侧翼的外显子中的+2e至-4e区域内;或在位于该保留内含子的3’剪接位点侧翼的外显子中的+2e至-4e区域内。
在实施方案中,所述组合物的或如本文所述方法中所用的反义寡聚体并不是通过调节由编码靶蛋白或功能rna的基因所转录的前mrna的其它剪接来增加该靶蛋白或功能rna的量。在实施方案中,所述组合物的或如本文所述方法中所用的反义寡聚体并不是通过调节由编码靶蛋白或功能rna的基因的突变导致的异常剪接来增加该靶蛋白或功能rna的量。
在实施方案中,所述ric前mrna由全长前mrna或野生型前mrna的部分剪接而产生。在实施方案中,编码靶蛋白或功能rna的mrna是全长成熟mrna或野生型成熟mrna。在实施方案中,所产生的靶蛋白是全长蛋白或野生型蛋白。在实施方案中,所产生的功能rna是全长功能rna或野生型功能rna。
在实施方案中,所述保留内含子为限速内含子。在实施方案中,该保留内含子为所述ric前mrna中最丰富的内含子。在实施方案中,该保留内含子为所述ric前mrna中第二丰富的内含子。
在实施方案中,所述组合物的或如本文所述方法中所用的反义寡聚体包含主链修饰,该主链修饰包含硫代磷酸酯键或磷二酰胺键。在实施方案中,该反义寡聚体为反义寡核苷酸。
在实施方案中,该反义寡聚体包含磷二酰胺吗啉基、锁定核酸、肽核酸、2’-o-甲基、2’-氟或2’-o-甲氧乙基部分。在实施方案中,该反义寡聚体包含至少一个经修饰的糖部分。在相关的实施方案中,每一个糖部分均为经修饰的糖部分。
该反义寡聚体可由8至50个核碱基组成。在实施方案中,反义寡聚体由8至40个核碱基、8至35个核碱基、8至30个核碱基、8至25个核碱基、8至20个核碱基、8至15个核碱基、9至50个核碱基、9至40个核碱基、9至35个核碱基、9至30个核碱基、9至25个核碱基、9至20个核碱基、9至15个核碱基、10至50个核碱基、10至40个核碱基、10至35个核碱基、10至30个核碱基、10至25个核碱基、10至20个核碱基、10至15个核碱基、11至50个核碱基、11至40个核碱基、11至35个核碱基、11至30个核碱基、11至25个核碱基、11至20个核碱基、11至15个核碱基、12至50个核碱基、12至40个核碱基、12至35个核碱基、12至30个核碱基、12至25个核碱基、12至20个核碱基或12至15个核碱基组成。
在实施方案中,该反义寡聚体与编码所述蛋白质的ric前mrna的靶向部分至少80%、至少85%、至少90%、至少95%、至少98%、至少99%或100%互补。在实施方案中,该反义寡聚体与选自seqidno:1-102的ric前mrna的一部分结合。
在实施方案中,该反义寡聚体被包含在包含赋形剂的药物组合物中。
本文描述了从每一个均与编码靶蛋白或功能rna的ric前mrna的靶区域杂交的一组反义寡聚体中鉴定反义寡聚体的方法,该反义寡聚体通过诱导保留内含子从所述ric前mrna中组成性剪接来增加编码该靶蛋白或功能rna的mrna的量,其中所述ric前mrna包含至少一个保留内含子,其中该组中的反义寡聚体以每1至5个核苷酸进行平铺(tile),并且其中该组中的反义寡聚体与在以下序列内的ric前mrna杂交,该序列在:所述至少一个保留内含子的5’剪接位点上游约100个核苷酸至所述至少一个保留内含子的5’剪接位点下游约100个核苷酸;或所述至少一个保留内含子的3’剪接位点上游约100个核苷酸至所述至少一个保留内含子的3’剪接位点下游约100个核苷酸;所述方法包括:a)将该组中的第一反义寡聚体递送至包含该ric前mrna的细胞;b)在第一反义寡聚体所递送到的细胞中测量ric前mrna的量和测量编码该靶蛋白或功能rna的mrna的量;c)在对照细胞中测量ric前mrna的量和测量编码靶蛋白或功能rna的mrna的量;以及d)比较在b和c中所测得的ric前mrna和编码靶蛋白或功能rna的mrna的量;其中根据与对照细胞相比,在第一反义寡聚体所递送到的细胞中观察到ric前mrna的量减少和观察到编码靶蛋白或功能rna的mrna的量增加,将第一反义寡聚体鉴定为通过诱导所述至少一种保留内含子从ric前mrna中组成性剪接而增加编码该靶蛋白或功能rna的mrna的量的反义寡聚体;以及根据需要采用该组反义寡聚体中另外的反义寡聚体来重复步骤a到d,以鉴定通过诱导保留内含子从ric前mrna中组成性剪接而增加细胞中来自基因的mrna的量的反义寡聚体。
本文还描述了鉴定用于治疗病况的反义寡聚体(aso)的方法,其中该病况由基因产物的产生量不足所导致,该方法包括:鉴定来自具有该病况的受试者的细胞核中至少一种ric前mrna的存在,其中该ric前mrna包含至少一个保留内含子且由编码该基因产物的基因转录,并且其中所鉴定的ric前mrna在完全剪接为成熟mrna时编码完全功能性或部分功能性形式的基因产物;a)制备每一个均与所述至少一个ric前mrna的靶区域杂交的一组aso,其中该组中的反义寡聚体以每1至5个核苷酸进行平铺,并且其中该组中的反义寡聚体与在以下序列内的至少一种ric前mrna杂交,该序列在:所述至少一个保留内含子的5’剪接位点上游约100个核苷酸至所述至少一个保留内含子的5’剪接位点下游约100个核苷酸;或所述至少一个保留内含子的3’剪接位点上游约100个核苷酸至所述至少一个保留内含子的3’剪接位点下游约100个核苷酸;b)将该组aso中的第一aso递送至包含所述至少一个ric前mrna的细胞;c)在第一反义寡聚体所递送到的细胞中测量ric前mrna的量和测量编码该基因产物的mrna的量;d)在对照细胞中测量ric前mrna的量和测量编码该基因产物的mrna的量;以及e)比较步骤c和d中得到的值;其中根据与对照细胞相比,在第一反义寡聚体所递送到的细胞中观察到ric前mrna的量减少和观察到编码该基因产物的mrna的量增加,将第一反义寡聚体鉴定为通过诱导所述至少一种保留内含子从ric前mrna中组成性剪接而增加编码该基因产物的mrna的量的反义寡聚体;以及根据需要采用该组反义寡聚体中另外的反义寡聚体来重复步骤a到e,以鉴定通过诱导保留内含子从ric前mrna中组成性剪接而增加编码细胞中来自基因的基因产物的mrna的量的反义寡聚体;以及对于通过诱导保留内含子从ric前mrna中组成性剪接而增加细胞中编码所述基因产物的mrna的量的反义寡聚体,进一步测试其增加由细胞产生的基因产物的量的能力。
援引并入
本说明书中提及的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别且单独地指出每一个单独的出版物、专利或专利申请通过引用并入。
附图说明
并未打算按比例来绘制附图。附图仅是说明性的并且不是实现本发明所需的。出于清楚的目的,可以不将每一个组分都标在每张图中。在所述附图中:
图1示出了示例性的含保留内含子(ric)的前mrna转录物的示意图。用-3e至-1e和+1至+6(标有“e”的数字是外显子的而未标记的数字是内含子的)的加下划线字母(字母为核苷酸;大写字母:外显子部分,小写字母:内含子部分)指示5’剪接位点共有序列。用-15至-1和+1e(标有“e”的数字是外显子的而未标记的数字是内含子的)的加下划线字母(字母为核苷酸;大写字母:外显子部分,小写字母:内含子部分)指示3’剪接位点共有序列。用于aso筛选的内含子靶区域包含相对于保留内含子的5’剪接位点(左侧箭头)的+6至相对于保留内含子的3’剪接位点(右侧箭头)的-16的核苷酸。在实施方案中,用于aso筛选的内含子靶区域包含相对于保留内含子的5’剪接位点的+6至+100和相对于保留内含子的3’剪接位点的-16至-100的核苷酸。外显子靶区域包含位于保留内含子的5’剪接位点侧翼的外显子中的+2e至-4e和位于保留内含子的3’剪接位点侧翼的外显子中的+2e至-4e的核苷酸。“n”或“n”表示任何核苷酸,“y”表示嘧啶。所示出的序列代表哺乳动物剪接位点的共有序列,并且单独的内含子和外显子不需要在每个位置匹配该共有序列。
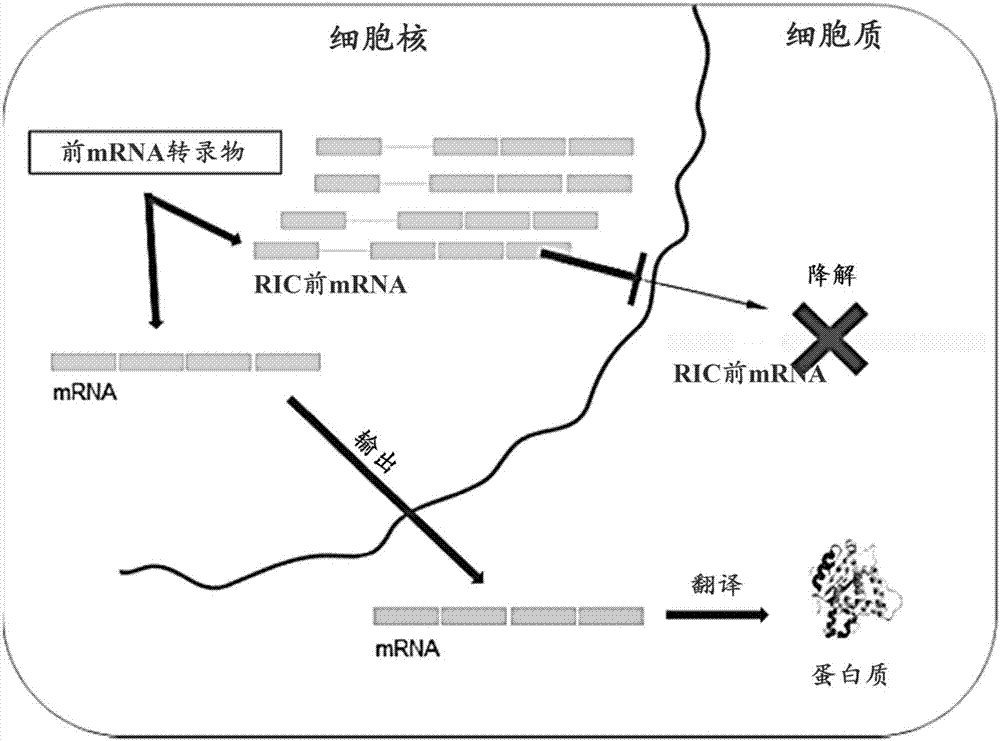
图2a-2b示出了核基因输出定向增加(tango)方法的示意图。图2a显示了分为细胞核和细胞质区室的细胞。在细胞核中,由外显子(矩形)和内含子(连接线)组成的靶基因的前mrna转录物经历剪接以生成mrna,并且该mrna被输出到细胞质并翻译为靶蛋白。对于该靶基因,内含子1的剪接是低效的且含保留内含子(ric)的前mrna主要在细胞核中积累,并且如果输出到细胞质则降解,导致无靶蛋白产生。图2b示出了分为细胞核和细胞质区室的相同细胞的实例。用反义寡聚体(aso)处理促进了内含子1的剪接并导致mrna的增加,该增加的mrna继而翻译为更高水平的靶蛋白。
图3示出了利用如实施例1所述的rt-pcr筛选7-外显子/6-内含子基因的内含子保留的实例的示意图。编号矩形表示通过表示内含子的线连接的外显子。弓形箭头指示剪接事件。短的水平条表示用来评估内含子保留的引物对。正向引物用“f”指示而反向引物则用“r”指示,即,f1和r1、f2和r2、f3和r3、f4和r4、f5和r5以及f6和r6对。当这样的内含子存在且观察到相邻内含子被剪接掉(去除)时,该内含子被鉴定为保留内含子。
图4示出了利用如实施例2所述的rt-pcr筛选以确认7-外显子/6-内含子基因的内含子保留的实例的示意图。编号矩形表示通过表示内含子的线连接的外显子。弓形箭头指示剪接事件。短的水平条表示用来评估内含子保留的引物对。正向引物用“f”标记而反向引物则用“r1”、“r2”或“r3”标记。当这样的内含子存在且观察到一个或多个相邻内含子被剪接掉(去除)时,所述内含子被鉴定为保留内含子。
图5示出了测定内含子去除效率的示例性核糖核酸酶保护测定(rpa)的示意图。
图6a-6e示出了如实施例1所述鉴定prpf31和rb1基因中的内含子保留事件。图6a示出了prpf31基因的示意图,其中编号矩形表示外显子且中间连线表示内含子。正向(“f”)和反向(“r”)引物由短线表示。下面是示出对应于prpf31中内含子保留事件的rt-pcr产物的代表性凝胶。该产物在1.5%溴化乙锭染色的琼脂糖凝胶中分离。顶部凝胶对应于来自hela细胞的细胞核部分的产物,而底部凝胶则对应于来自293t细胞的细胞核部分的产物。星号指示对于内含子保留事件的正确产物(按大小)。图6b示出了rb1基因的示意图,其中编号矩形表示外显子且中间连线表示内含子。下面是显示对应于rb1中内含子保留事件的来自hela细胞核提取物的rt-pcr产物的代表性凝胶。该rt-pcr产物在1.5%溴化乙锭染色的琼脂糖凝胶中分离。图6c示出了对应于rb1中内含子保留事件的来自293t细胞核提取物的rt-pcr产物的代表性凝胶。图6d示出了对应于prpf31和rb1中内含子保留事件的来自arpe-19细胞核提取物的rt-pcr产物的代表性凝胶。rt-pcr产物在1.5%溴化乙锭染色的琼脂糖凝胶中分离。图6e示出了对应于prpf31和rb1中内含子保留事件的来自arpe-19细胞质提取物的rt-pcr产物的代表性凝胶。ivs:间插序列(内含子)。
图7a-7b示出了如实施例2所述鉴定prpf31和rb1基因中的内含子保留事件。图7a示出了对应于prpf31中内含子保留事件的rt-pcr产物的代表性凝胶。来自arpe-19细胞核提取物的rt-pcr产物在1.5%溴化乙锭染色的琼脂糖凝胶中分离。图7b示出了对应于rb1中内含子保留事件的rt-pcr产物的代表性凝胶。来自arpe-19细胞核提取物的rt-pcr产物在1.5%溴化乙锭染色的琼脂糖凝胶中分离。星号指示利用所示引物对针对内含子保留事件的正确产物(按大小)。ivs:间插序列(内含子)。
图8a-8c示出了如实施例3所述通过经由剪接位点诱变提升剪接效率而增加的基因表达。图8a示出了hbb报道基因的示意图,其包括表示外显子的编号矩形。绘制标记出内含子-外显子边界的实际hbb剪接位点序列。用星号指示的剪接位点序列内的核苷酸显示通过将剪接位点序列带入共有序列(在hbb剪接位点正下方的序列)的定点诱变而引入的核苷酸置换的位置。a:ivs15’剪接位点突变体,b:ivs13’剪接位点突变体,c:ivs25’剪接位点突变体,d:ivs23’剪接位点突变体。ab、cd、ac和bd:组合突变体。图8b示出了野生型(wt)和突变hbb报道基因的放射性rt-pcr产物的代表性凝胶。该rt-pcr产物在5%聚丙烯酰胺凝胶中分离。图8c示出了相对于gfp归一化的对应于hbb转录物的条带强度的柱状图。以相对于wthbb产物的倍数变化进行绘图。黑线指示比率为1,没有变化。
图9a-9c示出了如实施例3所述,靶向hbbivs15’剪接位点下游紧邻序列的aso增加了hbbmrna。图9a示出了hbb报道基因的示意图。编号矩形表示外显子,并且中间连线表示内含子。橙色线指示ivs1+6aso(“+6”),灰色线指示ivs1+7aso(“+7”)。黑色线指示在hbb转录物的pcr扩增中使用的正向(“f”)和反向(“r”)引物。图9b呈现了未处理的(-)、模拟处理的(rim、rnaimax或ep、endoporter)或用所示浓度的非靶向(nt)或ivs1+72′-o-me(凝胶的左侧部分)或pmo(凝胶的右侧部分)aso处理的野生型hbb报道基因的放射性rt-pcr产物的代表性凝胶。该rt-pcr产物在5%聚丙烯酰胺凝胶中分离。图9c示出了相对于gfp归一化的对应于hbb转录物的条带强度的柱状图。以相对于来自模拟处理的细胞的产物的倍数变化进行绘图。绿柱对应于用ivs+72′-o-measo处理,而橙柱对应于用ivs+7pmoaso处理。黑线指示比率为1,没有变化。
图10a-10c示出了如实施例4所述,靶向hbbivs15’剪接位点下游紧邻序列的ivs1+72′-o-measo增加了gfp-hbb-t7蛋白水平。图10a示出了已整合在u2os细胞的基因组中的gfp-hbb-t7报道基因的示意图。标有“gfp”的矩形表示gfp的开放读框,编号矩形表示hbb外显子,中间连线表示内含子,而标有“t7”的矩形表示编码t7标签的序列。标有“+7”的线指示ivs1+7aso。图10b呈现了模拟处理的(rim、rnaimax)或用浓度为50nm的ivs1+72′-o-measo处理的野生型gfp-hbb-t7报道基因的蛋白质产物的代表性凝胶。该蛋白质产物在4-20%sds-聚丙烯酰胺凝胶上分离。采用抗gfp和β微管蛋白的抗体来检测蛋白质产物。图10c示出了来自两个生物复制品的相对于β微管蛋白归一化的对应于gfp-hbb-t7蛋白的条带强度的柱状图。以相对于来自模拟处理的细胞的产物的倍数变化进行绘图。黑线指示比率为1,没有变化。
图11示出了如实施例5所述,在ucsc基因组浏览器中所显现的,利用rna测序(rnaseq)鉴定adamts13基因中的内含子保留事件。顶部图幅(panel)示出了对应于在thle-3(人肝上皮)细胞中表达且位于细胞质(顶部)或细胞核部分(底部)中的adamts13转录物的读数密度。在该幅图的底部,按比例示出了adamts13基因的所有参考序列(refseq.)同种型的图示。读数密度显示为峰。最高的读数密度对应于外显子(黑框),而对于任一细胞部分中的大多数内含子(带箭头的线)均未观察到读数。与细胞质部分相比,对于细胞核部分中的内含子25和27检测到较高的读数密度(箭头所指),这表明内含子25和27的剪接效率低,导致了内含子保留。含保留内含子的前mrna转录物保留在细胞核中而不向外输出到细胞质中。底部的图片详细地示出了对于thle-3细胞中的内含子25的读数密度。
图12示出了如实施例6所述,经由放射性rt-pcr验证生物信息学分析。图12描绘了验证图11所示生物信息学预测的放射性rt-pcr测定的示意图。编号矩形表示外显子,并且中间连线表示内含子。黑线指示在adamts-13转录物的pcr扩增中使用的正向(“f1”)和反向(“r1”)引物,其产生两种产物:内含子25保留的(652bp)和正确剪接的(187bp)产物。下方示出在5%聚丙烯酰胺凝胶中分离的来自a172(成胶质细胞瘤,左侧)和hepg2(肝细胞癌,右侧)细胞的细胞核和细胞质部分的放射性rt-pcr产物的代表性凝胶。星号指示正确的产物(按大小)。结果显示了对应于两个细胞系的细胞核部分中的内含子25保留的产物的条带,该产物不存在于两个细胞质部分中。右侧表格中则示出了根据放射性rt-pcr和rnaseq实验按内含子保留百分比(pir)计算的adamts13内含子25保留的量化总结。
图13示出了如实施例7所述,利用2′-o-measo(ps主链),针对5’剪接位点下游紧邻或3’剪接位点上游紧邻的adamts13ivs25靶向序列进行的aso步移(walk)的图示。将aso设计为通过一次移位5个核苷酸来覆盖这些区域。按比例绘制外显子24至29和内含子序列。
图14描绘了如实施例8所述的靶向内含子25的aso步移的结果。在顶部,代表性凝胶示出了hepg2细胞中用浓度为60nm的模拟处理的(-,仅rnaimax)、经smn-对照aso处理的或如图13所述靶向内含子25的2′-o-measo处理的adamts13的放射性rt-pcr产物。来自3个独立实验的相对于β肌动蛋白归一化的对应于adamts13产物的条带的量化以相对于smn-对照-aso处理的产物的倍数变化绘制在下面的柱状图中。黑线指示比率为1,没有变化。星号指示导致mrna水平增加最多的aso。
图15示出了如实施例9所述,adam-ivs25+21、adam-ivs25+26(两种最佳靶向aso)以及adam-ivs-46(一种导致adamts13转录物减少的aso)的剂量-响应曲线。在顶部图幅中,代表性凝胶显示了来自模拟处理的、所示浓度的smn-对照、adam-ivs25+21、adam-ivs25+26或adam-ivs-46处理的hepg2细胞的放射性rt-pcradamts13产物。该rt-pcr产物在5%聚丙烯酰胺凝胶中分离。相对于β肌动蛋白归一化的对应于adamts13产物的条带的量化以相对于模拟处理的产物的倍数变化绘制在下面的柱状图中。黑线指示比率为1,没有变化。
图16图示了如实施例10所述,用adam-ivs25+21和adam-ivs25+26aso处理hepg2细胞而导致的adamts13蛋白的增加。在顶部图幅中,代表性凝胶显示了来自模拟处理的、所示浓度的adam-ivs25+21或adam-ivs25+26处理的hepg2细胞的adamts13蛋白产物。该蛋白质产物在8%sds-聚丙烯酰胺凝胶上分离。采用抗adamts-13和α微管蛋白的抗体来检测蛋白质产物。下方的柱状图示出了相对于α微管蛋白归一化的对应于来自adam-ivs25+21处理的细胞的adamts-13蛋白水平的条带强度的量化。以相对于来自模拟处理的细胞的产物的倍数变化进行绘图。黑线指示比率为1,没有变化。adam-ivs25+21以剂量依赖性的方式增加adamts13蛋白产物。
图17示出了如实施例11所述,利用2′-o-me、5’-me-胞嘧啶aso,针对在adam-ivs25+21和adam-ivs25+26aso的区域中的adamts13ivs25靶向序列进行的aso微步移(microwalk)的图示。将aso设计为通过一次移位1个核苷酸来覆盖该区域。按比例绘制外显子24至29和内含子序列。
图18描绘了如实施例12所述,靶向内含子25中adam-ivs25+21和adam-ivs25+26区域的aso微步移的结果。在顶部,代表性凝胶示出了hepg2中用浓度为60nm的模拟处理的(-)、经smn-对照aso处理的或2′-o-me、5’-me-胞嘧啶aso(如图17描述)处理的adamts13的放射性rt-pcr产物。来自2个独立实验的相对于β肌动蛋白归一化的对应于adamts13产物的条带的量化以相对于模拟处理的产物的倍数变化绘制在下方的柱状图中。黑线指示比率为1,没有变化。两个淡灰色的柱指示图14和图15中描述的ivs252′-o-measoadam-ivs25+21和adam-ivs25+26。
图19示出了如实施例13所述,在ucsc基因组浏览器中显现的,利用rna测序(rnaseq)来鉴定tsc1基因中的内含子保留事件。顶部图幅示出了对应于在hcn(原代人皮质神经元)细胞中表达且位于细胞质(顶部)或细胞核部分(底部)中的tsc1转录物的读数密度。在该幅图的底部,按比例示出了tsc1基因的所有参考序列同种型的图示。读数密度显示为峰。最高的读数密度对应于外显子(黑框),而对于任一细胞部分中的大多数内含子(带箭头的线)均未观察到读数。与细胞质部分相比,对于细胞核部分中的内含子5、10和11检测到较高的读数密度(箭头所指),这表明内含子5、10和11的剪接效率低,导致了内含子保留。含保留内含子的前mrna转录物保留在细胞核中而不向外输出到细胞质中。在底部图片中则详细示出了对于hcn细胞和ast(原代人星形细胞)细胞的内含子10的读数密度。
图20示出了如实施例14所述验证图19中所示生物信息学预测的放射性rt-pcr测定的示意图。编号矩形表示外显子,并且中间连线表示内含子。黑线指示在tsc1转录物的pcr扩增中使用的正向(“f1”)和反向(“r1”)引物,其产生两种产物:内含子10保留的(617bp)和正确剪接的(278bp)产物。下方是显示在5%聚丙烯酰胺凝胶中分离的来自a172(成胶质细胞瘤)细胞的细胞核和细胞质部分的放射性rt-pcr产物的代表性凝胶。结果示出了对应于a172细胞的细胞核部分中的内含子10保留的产物的条带,该产物在细胞质部分中显著减少。条带的量化表明约36%的tsc1转录物含有内含子10并且表明该产物保留在细胞核中。此外,如对于adamts13所示,放射性rt-pcr结果验证了生物信息学预测。右侧表格示出了根据放射性rt-pcr和rnaseq实验按内含子保留百分比(pir)计算的tsc1内含子10保留的量化总结。
图21示出了如实施例15所述,利用2′-o-measo(ps主链),针对5’剪接位点下游紧邻或3’剪接位点上游紧邻的tsc1ivs10靶向序列进行的aso步移的图示。将aso设计为通过一次移位5个核苷酸来覆盖这些区域。按比例绘制tsc1外显子-内含子结构。
图22描绘了如实施例16所述的靶向内含子10的aso步移的结果。在顶部,代表性凝胶示出了a172细胞中用浓度为60nm的模拟处理的(-)、经smn-对照aso处理的或如图21所述靶向内含子10的2′-o-measo处理的tsc1的放射性rt-pcr产物。来自2个独立实验的相对于β肌动蛋白归一化的对应于tsc1产物的条带的量化以相对于模拟处理的产物的倍数变化绘制在下方的柱状图中。黑线指示比率为1,没有变化。星号指示导致tsc1mrna水平增加的aso。
图23示出了如实施例17所述针对tsc1-ivs10+31aso的剂量-响应曲线。在顶部图幅中,代表性凝胶示出了来自模拟处理的、所示浓度的smn-对照或tsc1-ivs10+31处理的a172细胞的放射性rt-pcrtsc1产物。该rt-pcr产物在5%聚丙烯酰胺凝胶中分离。相对于β肌动蛋白归一化的对应于tsc1产物的条带的量化以相对于模拟处理的产物的倍数变化绘制在左下方的柱状图中。相对于模拟处理的产物的相同实验的rt-qpcr结果绘制在右侧柱状图上,以证实放射性rt-pcr结果。黑线指示比率为1,没有变化。
图24图示了如实施例18所述,用tsc1-ivs10+31aso处理a172细胞而导致的tsc1蛋白的增加。在顶部图幅中,代表性凝胶示出了来自模拟处理的、所示浓度的smn-对照或tsc1-ivs10+31aso处理的a172细胞的tsc1蛋白产物。该蛋白产物在10%sds-聚丙烯酰胺凝胶上分离。采用抗tsc1和α微管蛋白的抗体来检测该蛋白产物。下方的柱状图示出了相对α微管蛋白归一化的对应于来自tsc1-ivs10+31处理的细胞的tsc1蛋白的条带强度的量化。以相对于来自模拟处理的细胞的产物的倍数变化进行绘图。黑线指示比率为1,没有变化。tsc1-ivs10+31增加了tsc1蛋白产物。
图25示出了如实施例19所述,在ucsc基因组浏览器中显现的,采用rna测序(rnaseq)鉴定impdh1基因中的内含子保留事件。顶部图幅示出了对应于在arpe19(人视网膜上皮)细胞中表达且位于细胞质(顶部)或细胞核部分(底部)中的impdh1转录物的读数密度。在该幅图的底部,按比例示出了impdh1基因的所有参考序列同种型的图示。读数密度显示为峰。最高的读数密度对应于外显子(黑框),而对于任一细胞部分中的大多数内含子(带箭头的线)均未观察到读数。与细胞质部分相比,对于细胞核部分中的内含子14检测到较高的读数密度(箭头所指),这表明内含子14的剪接效率低,导致了内含子保留。含保留内含子的前mrna转录物保留在细胞核中而未向外输出到细胞质。在底部图片中详细示出了对于arpe19细胞的内含子14的读数密度。
图26示出了使用如实施例20所述的2′-o-measo(ps主链),针对5’剪接位点下游紧邻或3’剪接位点上游紧邻的impdh1ivs14靶向序列进行的aso步移的图示。将aso设计为通过一次移位5个核苷酸来覆盖这些区域,除非在aso中存在一段四个鸟嘌呤。按比例绘制impdh1外显子-内含子结构。
图27描绘了如实施例21所述的靶向内含子14的aso步移的结果。在顶部,代表性凝胶示出了arpe19细胞中用浓度为60nm的模拟处理的(-)、经smn-对照aso处理的或如图21所述靶向内含子14的2′-o-measo处理的impdh1的放射性rt-pcr产物。来自2个独立实验的相对于β肌动蛋白归一化的对应于impdh1产物的条带量化以相对于模拟处理的产物的倍数变化绘制在下方的柱状图中。黑线指示比率为1,没有变化。星号指示导致impdh1mrna水平增加最多的aso。
图28示出了如实施例22所述,用所示浓度的imp-ivs14+48aso处理arpe19细胞而导致的impdh1基因表达水平以剂量依赖性方式的增加。来自arpe-19细胞的impdh1(内含子14保留的和正确剪接的)和β肌动蛋白的放射性rt-pcr产物在5%聚丙烯酰胺凝胶上分离。左侧柱状图证实了与模拟处理的细胞相比,相对于来自imp-ivs14+48aso处理的细胞的总转录物(内含子14保留的和正确剪接的)所计算的内含子保留百分比(pir)的剂量依赖性减少(两个独立实验)。来自两个独立实验的正确剪接的转录物水平相对于模拟处理的细胞的倍数变化绘制在中间的图中,其示出了impdh1转录物水平的剂量依赖性增加。进行rt-qpcr(右侧柱状图)并将所得值相对于β肌动蛋白进行归一化。以四个生物复制品相对于模拟处理的impdh1产物的倍数变化进行绘图,确认了放射性rt-pcr结果。
图29示出了如实施例23所述,经由arpe19细胞中所示浓度的imp-ivs14+48aso靶向而获得的impdh1蛋白水平的增加。来自arpe-19细胞的蛋白质裂解物在4-20%sds-聚丙烯酰胺凝胶上分离。采用抗impdh1、β肌动蛋白和β连环蛋白的抗体来检测蛋白质产物。将impdh1蛋白条带的强度相对于β肌动蛋白条带的强度进行归一化,并计算相对于来自四个生物复制品的模拟处理的产物的倍数变化,并且绘制在下方的柱状图中。
图30示出了如实施例24所述,利用2′-o-me、5’-me-胞嘧啶aso,针对imp-ivs14+48aso的区域中的impdh1ivs14靶向序列进行的aso微步移的图示。将aso设计为通过一次移位1个核苷酸来覆盖该区域。按比例绘制impdh1外显子-内含子结构。
图31示出了如实施例25所述,如图30所示的微步移导致的mpdh1表达水平的增加。对从arpe-19细胞中提取的总rna进行rt–qpcr,所述总rna在60nm的aso浓度下进行处理。将impdh1基因的ct值相对于ct值β肌动蛋白(左侧)和rpl32(右侧)管家基因进行归一化,并将相对于来自模拟处理的细胞的产物的倍数变化绘制在柱状图中。该微步移鉴定了进一步增加impdh1转录物水平的两种另外的aso。
图32示出了如实施例26所述,在ucsc基因组浏览器中显现的,利用rna测序(rnaseq)鉴定pkd1基因中的内含子保留事件。顶部图幅示出了对应于在原代人肾上皮细胞(ren)中表达且位于细胞质(顶部)或细胞核部分(底部)中的pkd1转录物的读数密度。在该幅图的底部,按比例示出了pkd1基因的参考序列同种型的图示。读数密度示出为峰。最高的读数密度对应于外显子(黑框),而对于任一细胞部分中的大多数内含子(带箭头的线)均未观察到读数。与细胞质部分相比,对于细胞核部分中的内含子32、33、37和38检测到较高的读数密度(箭头所指),这表明这些内含子的剪接效率低,导致了内含子保留。含保留内含子的前mrna转录物保留在细胞核中而未向外输出到细胞质中。在底部图片中详细示出了对于ren细胞的内含子37的读数密度。
图33示出了如实施例27所述,利用2′-o-measo(ps主链),针对5’剪接位点下游紧邻或3’剪接位点上游紧邻的pkd1ivs37靶向序列进行的aso步移的图示。将aso设计为通过一次移位5个核苷酸来覆盖这些区域,除非在aso中存在一段四个鸟嘌呤。按比例绘制pkd1外显子-内含子结构。
图34描绘了如实施例28所述的靶向内含子37的aso步移的结果。在顶部,代表性凝胶示出了hek293(人胚肾上皮)细胞中用浓度为60nm的模拟处理的(c)、经smn-对照aso处理的或如图33所述靶向内含子37的2′-o-measo处理的pkd1的放射性rt-pcr产物。来自2个独立实验的相对于β肌动蛋白归一化的对应于pkd1产物的条带量化以相对于模拟处理的产物的倍数变化绘制在下方的柱状图中。黑线指示比率为1,没有变化。星号指示导致pkd1mrna水平增加最多的aso。
图35示出了如实施例29所述,利用所示浓度的pkd1-ivs37aso处理hek293细胞而导致的pkd1基因表达水平以剂量依赖性方式的增加。来自hek293细胞的pkd1(内含子37保留的和正确剪接的)和β肌动蛋白的放射性rt-pcr产物在5%聚丙烯酰胺凝胶上分离。左侧柱状图证实了与模拟处理的细胞相比,相对于来自pkd1-ivs37+29aso处理的细胞的总转录物(内含子37保留的和正确剪接的)所计算的内含子保留百分比(pir)的剂量依赖性减少(两个独立实验)。来自两个独立实验的正确剪接的转录物水平相对于模拟处理的细胞的倍数变化绘制在中间的图中,其示出了pkd1转录物水平的增加。进行rt-qpcr(右侧柱状图)并将所得值相对于β肌动蛋白进行归一化。以四个生物复制品相对于模拟处理的pkd1产物的倍数变化进行绘图,确认了放射性rt-pcr结果并且示出了pkd1转录物水平的剂量依赖性增加。
图36示出了如实施例30所述,经由hek293细胞中所示浓度的pkd1-ivs37+29aso靶向而获得的pkd1蛋白水平的增加。将hek293固定和透化处理,并用抗pkd1抗体或igg同种型对照进行处理。在每一种情况下针对10,000个处理的细胞进行流式细胞术分析,并对荧光强度进行绘图。计算相对于模拟处理的(未转染的)产物的倍数变化并将其绘制在下方的柱状图中,表明用pkd1-ivs37+29aso处理后pkd1水平的增加。
图37示出了如实施例31所述,在ucsc基因组浏览器中显现的,利用rna测序(rnaseq)鉴定ikbkap基因中的内含子保留事件。顶部图幅示出了对应于在arpe19、ast、原代人支气管上皮细胞(bron)、hcn、ren和thle3细胞中表达且位于细胞质(每一个细胞系的顶部)或细胞核部分(每一个细胞系的底部)中的pkd1转录物的读数密度。在该幅图的底部,按比例示出了ikbkap基因的所有参考序列同种型的图示。读数密度显示为峰。最高的读数密度对应于外显子(黑框),而对于任一细胞部分中的大多数内含子(带箭头的线)均未观察到读数。与细胞质部分相比,对于细胞核部分中的内含子7和8检测到较高的读数密度(箭头所指),这表明这些内含子的剪接效率低,导致了内含子保留。含保留内含子的前mrna转录物保留在细胞核中而未向外输出到细胞质中。在底部图中详细显示了对于所有分析的细胞的内含子7和8的读数密度。
图38分别示出了如实施例32所述,arpe-19、hela和u2os细胞系中的ikbkap内含子7保留水平。从arpe-19、hela和u2os细胞中提取细胞核和细胞质rna部分,并将它们相应的放射性rt-pcr产物在5%聚丙烯酰胺凝胶上分离。编号矩形表示外显子,并且中间连线表示内含子。结果示出了对应于三种细胞系的细胞核部分中的内含子7保留的产物的条带,该产物不存在于相应的细胞质部分中。条带的量化表明约35%的ikbkap转录物含有内含子7并且表明该产物保留在细胞核中。再者,放射性rt-pcr结果验证了生物信息学预测。右侧表格中显示了根据放射性rt-pcr以及rnaseq实验按相对于总转录物(内含子7保留的和正确剪接的)的内含子保留百分比(pir)计算的ikbkap内含子7保留的量化总结。
图39示出了如实施例33所述,利用2′-o-measo(ps主链),针对5’剪接位点下游紧邻或3’剪接位点上游紧邻的ikbkapivs7(顶部)和ivs8(底部)靶向序列进行的aso步移的图示。将aso设计为通过一次移位5个核苷酸来覆盖这些区域。按比例绘制ikbkap外显子-内含子结构。
图40示出了如实施例34所述,经由如图39所示内含子7(顶部)和8(底部)的特异性aso靶向而获得的ikbkap基因表达水平的增加。从用浓度为60nm的模拟处理的、smn-对照aso处理的或每一种aso处理的arpe-19细胞中提取细胞质rna。进行rt-qpcr以测量ikbkap表达水平,并将来自ikbkap的ct值相对于β肌动蛋白产物的相应ct值进行归一化。以相对于模拟物处理产物的倍数变化进行绘图。
图41表明如实施例35所述,在用所示浓度的ikb-ivs7+26或ikb-ivs8-16aso或用各为45nm(总计90nm)的两种aso的组合处理的细胞中,ikbkap转录物水平以剂量依赖性方式的增加。将利用来自arpe-19细胞的细胞质rna的对应于外显子6-8(ikb-ivs7+26,顶部)或外显子8-10(ikb-ivs8-16,底部)的放射性rt-pcr产物在5%聚丙烯酰胺凝胶上分离。ikbkap的表达通过测量条带强度予以量化,并将所述值相对于β-肌动蛋白的值进行归一化。以相对于模拟处理的细胞的产物的来自两个生物复制品的倍数变化进行绘图,并且将其显示在每个代表性凝胶右侧的柱状图中。
图42示出了如实施例36所述,在用所示浓度的ikb-ivs7+26或ikb-ivs8-16aso或用各为45nm(总计90nm)的两种aso的组合处理的arpe19细胞中,ikap蛋白水平的剂量依赖性增加。从arpe-19细胞中提取蛋白质裂解物并在4-20%sds-聚丙烯酰胺凝胶上分离。采用抗ikap和β连环蛋白的抗体来检测分离的蛋白质产物。将ikap蛋白条带的强度相对于β连环蛋白条带的强度进行归一化,并相对于模拟处理的细胞计算两个生物复制品的倍数变化,并绘制在下方的柱状图中。
序列
本申请包括核苷酸序列seqidno:1-374,并且这些核苷酸序列在权利要求书之前的表2至表8和表11至表20中列出。表11至表20中以seqidno:1-102示出的核苷酸序列是可被反义寡聚体通过本文所述方法予以靶向的序列的实例。表2至表8中以seqidno:103-374示出的核苷酸序列是可用于本文所述方法的反义寡聚体的实例。在所有的表格中,大写字母代表外显子序列而小写字母代表内含子序列。
具体实施方式
百分之八十五(85%)的人类蛋白质编码基因具有至少一个内含子;每个基因的内含子的平均数目为八,且内含子数目可在1至316的范围内。单独的内含子以不同的效率从初级转录物中剪接,并且在大多数情况下只有完全剪接的mrna才通过核孔输出以供随后在细胞质中翻译。在细胞核中可检测到未剪接的及部分剪接的转录物。一般认为,未完全剪接的转录物的细胞核保留是防止细胞质中可翻译为蛋白质的潜在有毒mrna的积累的机制。对于一些基因,在细胞质中翻译之前,最低效的内含子的剪接是在基因表达中的转录后限速步骤。如果对于基因表达的细胞核阶段限速的内含子剪接可更有效地进行,则完全剪接的成熟mrna的稳态产生量以及相应蛋白质的翻译可增加。此类方法还将帮助上调靶基因的表达,它具有大量临床和研究应用。增加基因的输出(正常和/或突变等位基因)可用于补偿减少其基因产物例如蛋白质或功能rna的活性量的任何突变。许多遗传病和遗传失调是蛋白质产生量减少或产生仅具有部分功能的蛋白质的结果。
如本文所用,术语“包含”或其变化形式如“包括”或“含有”应理解为指示包含任何所述特征(例如在反义寡聚体的情况下,包含限定的核碱基序列),但不排除任何其他特征。因此,如本文所用,术语“包含”是包含性的,并且不排除另外的未引用的特征(例如,在反义寡聚体的情况下,存在其他未述及的核碱基)。
在本文提供的任何组合物和方法的实施方案中,“包含”可以用“基本上由...组成”或“由...组成”代替。本文采用短语“基本上由...组成”来表示需要指定的特征(例如,核碱基序列)以及实质上不影响要求保护的发明的特性或功能的那些特征。如本文所用,术语“由...组成”用来指示仅存在所述特征(例如,核碱基序列)(因此在由指定核碱基序列组成的反义寡聚体的情况下,排除其他未述及的核碱基的存在)。
核基因输出的定向增加
本文描述了增加靶蛋白的表达的方法,其被称为核基因输出的定向增加(tango)。该方法包括使具有(包含)含保留内含子的前mrna(ric前mrna)的细胞与互补于ric前mrna的靶向部分的反义寡聚体(aso)相接触,该ric前mrna包含保留内含子,位于5’剪接位点侧翼的外显子,位于3’剪接位点侧翼的外显子,并且编码靶蛋白。aso与ric前mrna的所述部分的杂交导致在保留内含子的剪接位点(5’剪接位点或3’剪接位点)处增强的剪接,并且随后增加了靶蛋白产生量。
术语“前mrna”和“前mrna转录物”可互换使用,是指含有至少一个内含子的任何前mrna种类。前mrna或前mrna转录物可包含5’-7-甲基鸟苷帽和/或聚a尾部。在一些实施方案中,前mrna转录物不包含5’-7-甲基鸟苷帽和/或聚a尾部。前mrna转录物在未被翻译为蛋白质(或从细胞核转运到细胞质中)时为非生产性的信使rna(mrna)分子。
如本文所用,“含保留内含子的前mrna”(“ric前mrna”)是含有至少一个保留内含子的前mrna转录物。该ric前mrna含有保留内含子、位于该保留内含子的5’剪接位点侧翼的外显子、位于该保留内含子的3’剪接位点侧翼的外显子,并且该ric前mrna编码靶蛋白。“ric前mrna编码靶蛋白”被理解为当完全剪接时编码靶蛋白。“保留内含子”是当由相同基因编码的一个或多个其他内含子如相邻内含子已经从同一前mrna转录物中剪接掉时,存在于该前mrna转录物中的任何内含子。在一些实施方案中,保留内含子为编码靶蛋白的ric前mrna中最丰富的内含子。在实施方案中,保留内含子为细胞中编码靶蛋白的基因所转录的一群ric前mrna中最丰富的内含子,其中该组ric前mrna包含两个或更多个保留内含子。在实施方案中,靶向编码靶蛋白的ric前mrna群中最丰富的内含子的反义寡聚体诱导将该群中两个或更多个保留内含子剪接掉,包括该反义寡聚体靶向或结合的保留内含子。在实施方案中,由此产生编码靶蛋白的成熟mrna。术语“成熟mrna”和“完全剪接的mrna”在本文可互换使用,以描述编码靶蛋白的完全加工的mrna(例如,从细胞核输出到细胞质中并翻译为靶蛋白的mrna)或完全加工的功能rna。术语“生产性mrna”还可以用来描述编码靶蛋白的完全加工的mrna。
在一些实施方案中,所靶向的区域在保留内含子中,该保留内含子是编码靶蛋白的ric前mrna中第二丰富的内含子。例如,由于第二丰富的保留内含子的核苷酸序列的独特性,将aso设计为靶向特定核苷酸序列的便利性,和/或用aso靶向内含子导致的蛋白质产生的增量,所以可以靶向第二丰富的保留内含子而非最丰富的保留内含子。在实施方案中,该保留内含子为细胞中编码靶蛋白的基因所转录的ric前mrna群中第二丰富的内含子,其中该群ric前mrna包含两个或更多个保留内含子。在实施方案中,靶向编码靶蛋白的该群ric前mrna中第二丰富的内含子的反义寡聚体诱导将该群中的两个或更多个保留内含子剪接掉,包括该反义寡聚体靶向或结合的保留内含子。在实施方案中,由此产生编码靶蛋白的完全剪接(成熟)的mrna。
在实施方案中,反义寡聚体与ric前mrna中非保留内含子内的靶向区域互补。在实施方案中,ric前mrna的靶向部分在相对于该非保留内含子的5’剪接位点的+6至+100区域内;或在相对于该非保留内含子的3’剪接位点的-16至-100区域内。在实施方案中,ric前mrna的靶向部分在相对于该非保留内含子的5’剪接位点的+100至相对于该非保留内含子的3’剪接位点的-100区域内。在用来确定区域或序列的位置时,“在...内”被理解为包括所述位置处的残基。例如,+6至+100区域包括位置+6和+100处的残基。在实施方案中,由此产生编码靶蛋白的完全剪接(成熟)的mrna。
在一些实施方案中,ric前mrna的保留内含子是低效剪接的内含子。如本文所用,“低效剪接”可指与ric前mrna中另一剪接位点处的剪接频率相比,与保留内含子相邻的剪接位点(5’剪接位点或3’剪接位点)处相对低频率的剪接。术语“低效剪接”还可以指剪接位点处剪接的相对速率或动力学,其中“低效剪接的”内含子可以在与ric前mrna中另一内含子相比较慢的速率下被剪接或去除。
在一些实施方案中,在位于5’剪接位点侧翼的外显子的-3e至-1e和保留内含子的+1至+6处的9-核苷酸序列与相应的野生型序列相同。在一些实施方案中,在保留内含子的-15至-1和位于3’剪接位点侧翼的外显子的+1e处的16核苷酸序列与相应的野生型序列相同。如本文所用,“野生型序列”是指在ncbi生物和科学信息储存库(由nationalcenterforbiotechnologyinformation,nationallibraryofmedicine,8600rockvillepike,bethesda,mdusa20894运行)中登录的公开参考基因组中的靶基因的核苷酸序列。如本文所用,用“e”表示的核苷酸位置表示该核苷酸存在于外显子的序列中(例如,位于5’剪接位点侧翼的外显子或位于3’剪接位点侧翼的外显子)。
所述方法包括使细胞接触与编码靶蛋白或功能rna的前mrna的一部分互补的aso,从而导致靶蛋白或功能rna的表达增加。如本文所用,“接触”或施用于细胞是指提供与该细胞紧邻的aso从而使得aso与细胞相互作用的任何方法。与aso接触的细胞将该aso吸收或转运到该细胞中。所述方法包括使病况或疾病相关或病况或疾病有关的细胞与本文描述的任何aso相接触。在一些实施方案中,该aso可以进一步修饰或附接(例如,共价附接)到另一分子,以将该aso靶向至细胞类型,增强该aso与病况或疾病相关或病况或疾病有关的细胞之间的接触,或增强该aso的吸收。
如图2a所示,在细胞核中,由外显子和内含子组成的前mrna转录物经历剪接,以生成可从该细胞的细胞核输出到细胞质中的mrna,在细胞质中它被翻译为蛋白质。在含有至少一个低效剪接内含子(保留内含子)的前mrna转录物的实例中,存在ric前mrna,它被保持在细胞核中,并且如果它被输出到细胞质则不翻译为蛋白质而是降解。不希望受到任何特定理论的束缚,在与前mrna转录物的靶向部分互补的aso的存在下,保留内含子的剪接得到增强,从而也增加了可输出并翻译为蛋白质的mrna的量(图2b)。
如本文所用,术语“增加蛋白质产生量”或“增加靶蛋白的表达”意指增加细胞中由mrna翻译的蛋白质(例如,靶蛋白)的量。“靶蛋白”可以是需要增加表达/产生量的任何蛋白质。在一些实施方案中,该靶蛋白是疾病相关蛋白质,如表1中示出的任何蛋白质。例如,使表达ric前mrna的细胞接触与ric前mrna转录物的靶向部分互补的aso导致由该前mrna编码的蛋白质(例如,靶蛋白)的量有可测得的增加。测量或检测蛋白质的产生量的方法对于本领域技术人员是显而易见的,并且包括例如蛋白质印迹法、流式细胞术、免疫荧光显微术和elisa。
在一些实施方案中,使细胞接触与ric前mrna转录物的靶向部分互补的aso,与在不存在该aso/不存在处理的情况下由细胞产生的蛋白质的量相比,导致所产生的蛋白质(例如,靶蛋白)的量增加了至少10%、20%、30%、40%、50%、60%、80%、100%、200%、300%、400%、500%或1000%。在实施方案中,与对照化合物产生的靶蛋白的量相比,该反义寡聚体所接触的细胞产生的靶蛋白的总量增加至约1.1倍至约10倍、约1.5倍至约10倍、约2倍至约10倍、约3倍至约10倍、约4倍至约10倍、约1.1倍至约5倍、约1.1倍至约6倍、约1.1倍至约7倍、约1.1倍至约8倍、约1.1倍至约9倍、约2倍至约5倍、约2倍至约6倍、约2倍至约7倍、约2倍至约8倍、约2倍至约9倍、约3倍至约6倍、约3倍至约7倍、约3倍至约8倍、约3倍至约9倍、约4倍至约7倍、约4倍至约8倍、约4倍至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。对照化合物可以是例如不与ric前mrna的靶向部分互补的寡核苷酸。
在一些实施方案中,使细胞与互补于ric前mrna转录物的靶向部分的aso相接触导致编码靶蛋白或功能rna的mrna(包括编码该靶蛋白或功能rna的成熟mrna)的量增加。在一些实施方案中,与在不存在该aso/不存在处理的情况下由细胞所产生的蛋白质的量相比,编码靶蛋白或功能rna的mrna或编码靶蛋白或功能rna的成熟mrna的量增加了至少10%、20%、30%、40%、50%、60%、80%、100%、200%、300%、400%、500%或1000%。在实施方案中,与未处理的细胞例如未处理的细胞或用对照化合物处理的细胞中产生的成熟rna的量相比,在反义寡聚体所接触的细胞中产生的编码靶蛋白或功能rna的mrna或编码靶蛋白或功能rna的成熟mrna的总量增加至约1.1倍至约10倍、约1.5倍至约10倍、约2倍至约10倍、约3倍至约10倍、约4倍至约10倍、约1.1倍至约5倍、约1.1倍至约6倍、约1.1倍至约7倍、约1.1倍至约8倍、约1.1倍至约9倍、约2倍至约5倍、约2倍至约6倍、约2倍至约7倍、约2倍至约8倍、约2倍至约9倍、约3倍至约6倍、约3倍至约7倍、约3倍至约8倍、约3倍至约9倍、约4倍至约7倍、约4倍至约8倍、约4倍至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。对照化合物可以是例如不与ric前mrna的靶向部分互补的寡核苷酸。
在实施方案中,使细胞与互补于ric前mrna转录物的靶向部分的aso相接触导致功能rna的量增加。在一些实施方案中,与在不存在aso/不存在处理的情况下由细胞所产生的功能rna的量相比,功能rna的量增加了至少10%、20%、30%、40%、50%、60%、80%、100%、200%、300%、400%、500%或1000%。在实施方案中,与未处理的细胞例如未处理的细胞或用对照化合物处理的细胞中产生的功能rna的量相比,该反义寡聚体所接触的细胞中产生的功能rna的总量增加至约1.1倍至约10倍、约1.5倍至约10倍、约2倍至约10倍、约3倍至约10倍、约4倍至约10倍、约1.1倍至约5倍、约1.1倍至约6倍、约1.1倍至约7倍、约1.1倍至约8倍、约1.1倍至约9倍、约2倍至约5倍、约2倍至约6倍、约2倍至约7倍、约2倍至约8倍、约2倍至约9倍、约3倍至约6倍、约3倍至约7倍、约3倍至约8倍、约3倍至约9倍、约4倍至约7倍、约4倍至约8倍、约4倍至约9倍,至少约1.1倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约5倍或至少约10倍。对照化合物可以是例如不与ric前mrna的靶向部分互补的寡核苷酸。可利用本文提供的任何方法来增加功能rna,例如不编码蛋白质的mrna如非蛋白质编码rna的产生量。在一些实施方案中,功能rna或非蛋白质编码rna与病况例如疾病或病症相关。
保留内含子从ric前mrna中的组成性剪接
本文提供的方法和反义寡核苷酸组合物可用于通过增加编码靶蛋白或功能rna的mrna或编码靶蛋白或功能rna的成熟mrna的水平,来增加例如在具有由该靶蛋白或功能rna的量或活性缺陷引起的病况的受试者的细胞中靶蛋白或功能rna的表达。特别地,本文所述的方法和组合物诱导保留内含子从编码靶蛋白或功能rna的ric前mrna转录物中的组成性剪接,从而增加编码该靶蛋白或功能rna的mrna,或编码该靶蛋白或功能rna的成熟mrna的水平,并增加该靶蛋白或功能rna的表达。
保留内含子从ric前mrna中的组成性剪接将该保留内含子从该ric前mrna中正确地去除,其中该保留内含子具有野生型剪接序列。如本文所用,组成性剪接不包括保留内含子从由具有以下突变的基因或等位基因转录的ric前mrna中的剪接,该突变引起由该基因或等位基因转录的前mrna的其它剪接或异常剪接。例如,利用本文提供的方法和反义寡核苷酸诱导的保留内含子的组成性剪接并不校正前mrna中的异常剪接或并不影响前mrna的其它剪接,以导致靶蛋白或功能rna的表达增加。
在实施方案中,组成性剪接将保留内含子从ric前mrna中正确地去除,其中该ric前mrna由野生型基因或等位基因或编码完全功能性靶蛋白或功能rna的多态性基因或等位基因转录,并且其中该基因或等位基因不具有引起该保留内含子的其它剪接或异常剪接的突变。
在一些实施方案中,保留内含子从编码靶蛋白或功能rna的ric前mrna中的组成性剪接将保留内含子从编码该靶蛋白或功能rna的ric前mrna中正确地去除,其中该ric前mrna由基因或等位基因转录,该基因或等位基因与由野生型等位基因产生相比以降低的水平产生靶基因或功能rna,并且其中该基因或等位基因不具有引起该保留内含子的其它剪接或异常剪接的突变。在这些实施方案中,当与对应的野生型蛋白或功能rna相比时,组成性剪接的保留内含子的正确去除导致功能性的靶蛋白或功能rna的产生。
在其他实施方案中,组成性剪接将保留内含子从ric前mrna中正确地去除,其中该ric前mrna由基因或等位基因转录,该基因或等位基因编码与对应的野生型蛋白或功能rna相比以功能降低的形式产生的靶蛋白或功能rna,并且其中该基因或等位基因不具有引起该保留内含子的选择性剪接或异常剪接的突变。在这些实施方案中,当与对应的野生型蛋白或功能rna相比时,组成性剪接的保留内含子的正确去除导致部分功能性的靶蛋白或部分功能性的功能rna的产生。
通过组成性剪接“正确去除”保留内含子是指去除整个内含子,而不去除外显子的任何部分。
在实施方案中,本文描述的或在任何本文所述方法中使用的反义寡聚体并不是通过调节由编码功能rna或靶蛋白的基因转录的前mrna的其它剪接或异常剪接来增加编码该靶蛋白或功能rna的mrna的量、该靶蛋白的量或该功能rna的量。可以利用任何已知的分析rna种类的序列和长度的方法,例如,通过rt-pcr以及使用本文别处和文献中描述的方法,来测量其它剪接或异常剪接的调节。在实施方案中,根据所剪接的感兴趣种类的量增加或减少至少10%或至1.1倍来确定其它或异常剪接的调节。在实施方案中,如本文关于在本发明的方法中测定编码靶蛋白或功能rna的mrna的增加所描述的,根据增加或减少至少10%至100%或至1/1.1至1/10的水平来确定调节。
在实施方案中,所述方法是一种通过部分剪接野生型前mrna而产生ric前mrna的方法。在实施方案中,该方法是一种通过部分剪接野生型前mrna而产生ric前mrna的方法。在实施方案中,该ric前mrna由全长前mrna的部分剪接而产生。在这些实施方案中,与具有野生型剪接位点序列的保留内含子的剪接相比,全长前mrna可在保留内含子的剪接位点处具有不损害保留内含子的正确剪接的多态性。
在实施方案中,编码靶蛋白或功能rna的mrna是全长成熟mrna或野生型成熟mrna。在这些实施方案中,与由野生型成熟mrna编码的靶蛋白或功能rna的活性相比,全长成熟mrna可具有不影响由成熟mrna编码的靶蛋白或功能rna的活性的多态性。
反义寡聚体
本公开内容的一方面是包含通过与ric前mrna的靶向部分结合而加强剪接的反义寡聚体的组合物。如本文所用,术语“aso”和“反义寡聚体”可互换使用,并指包含核碱基的寡聚体如多核苷酸,该寡聚体通过watson-crick碱基配对或摆动碱基配对(g-u)与靶核酸(例如,ric前mrna)序列杂交。aso可具有互补于靶序列的确切序列或近似的互补性(例如,足以结合靶序列和加强剪接位点处的剪接的互补性)。将aso设计为使得它们与靶核酸(例如,前mrna转录物的靶向部分)结合(杂交)并在生理条件下保持杂交。通常,如果它们与预期(靶向)核酸序列之外的位点杂交,则它们与有限数目的非靶核酸的序列(一些非靶核酸的位点)杂交。aso的设计可以考虑前mrna转录物的靶向部分的核酸序列,或基因组或细胞前mrna或转录物组中其他位置中的足够类似的核酸序列的存在,使得aso将结合其他位点并引起“脱靶”效应的可能性有限。本领域已知的,例如在作为wo2015/035091公开的名称为“reducingnonsense-mediatedmrnadecay”的pct申请号pct/us2014/054151中的任何反义寡聚体可用来实施本文所述的方法。
在一些实施方案中,aso与靶核酸或ric前mrna的靶向部分“特异性杂交”或对其为“特异的”。通常,此类杂交的发生伴随着基本大于37℃,优选至少50℃,并且通常为60℃至约90℃的tm。这样的杂交优选对应于严格的杂交条件。在给定的离子强度和ph下,tm为50%的靶序列与互补寡核苷酸杂交时的温度。
当杂交在两个单链多核苷酸之间的反平行结构中发生时,寡聚体如寡核苷酸彼此“互补”。如果杂交可在第一多核苷酸的一条链与第二多核苷酸之间发生,则双链多核苷酸可与另一个多核苷酸“互补”。互补性(一个多核苷酸与另一个互补的程度)是按照在相对链中根据普遍接受的碱基配对原则预计彼此形成氢键的碱基的比例(例如,百分比)而可量化的。反义寡聚体(aso)的序列不必与其靶核酸的序列100%互补才能杂交。在某些实施方案中,aso可包含与其靶向的靶核酸序列内的靶区域至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少96%、至少97%、至少98%或至少99%的序列互补性。例如,寡聚体化合物的20个核碱基中的18个与靶区域互补并将因此特异性杂交的aso将表现出90%的互补性。在该实例中,其余非互补的核碱基可与互补的核碱基聚集在一起或散布,并且不必彼此相接或与互补的核碱基相接。常规地可利用blast程序(基本局部比对检索工具)和本领域已知的powerblast程序(altschul等人,j.mol.biol.,1990,215,403-410;zhang和madden,genomeres.,1997,7,649-656)来确定aso与靶核酸的区域的互补性百分比。
aso不需要与靶序列中的所有核碱基杂交,并且与之杂交的核碱基可以是连续的或不连续的。aso可以在前mrna转录物的一个或多个区段上杂交,使得介于中间或相邻的区段不参与该杂交事件(例如,可形成环结构或发夹结构)。在某些实施方案中,aso与靶前mrna转录物中不连续的核碱基杂交。例如,aso可以与前mrna转录物中被不与aso杂交的一个或多个核碱基隔开的核碱基杂交。
本文所述的aso包含与在ric前mrna的靶部分中存在的核碱基互补的核碱基。术语aso包括寡核苷酸以及任何其他包含能够与靶mrna上的互补核碱基杂交的核碱基但不包含糖部分如肽核酸(pna)的寡聚体分子。aso可包含天然存在的核苷酸、核苷酸类似物、修饰的核苷酸或前述两个或三个的任意组合。术语“天然存在的核苷酸”包括脱氧核糖核苷酸和核糖核苷酸。术语“修饰的核苷酸”包括具有修饰的或取代的糖基团和/或具有修饰的主链的核苷酸。在一些实施方案中,aso的所有核苷酸是修饰的核苷酸。与本文所述的方法和组合物相容的aso的化学修饰或aso的组分对于本领域技术人员将是显而易见的,并且可在例如第8,258,109b2号美国专利、第5,656,612号美国专利、第2012/0190728号美国专利公开以及dias和stein,mol.cancerther.2002,1,347-355中找到,其均通过引用它们的全文并入本文。
aso的核碱基可以是任何天然存在的、未修饰的核碱基如腺嘌呤、鸟嘌呤、胞嘧啶、胸腺嘧啶和尿嘧啶,或任何合成或修饰的核碱基,其十分类似于未修饰的核碱基,使得其与靶前mrna上存在的核碱基能够氢键键合。修饰的核碱基的实例包括但不限于次黄嘌呤、黄嘌呤、7-甲基鸟嘌呤、5,6-二氢尿嘧啶、5-甲基胞嘧啶和5-羟甲基胞嘧啶。
本文所述的aso还包含连接寡聚体的组分的主链结构。术语“主链结构”和“寡聚体连接”可互换使用并指该aso的单体之间的连接。在天然存在的寡核苷酸中,主链包含连接寡聚体的糖部分的3’-5’磷酸二酯键。本文所述的aso的主链结构或寡聚体连接可包括(但不限于)硫代磷酸酯(phosphorothioate)、二硫代磷酸酯(phosphorodithioate)、硒代磷酸酯(phosphoroselenoate)、二硒代磷酸酯(phosphorodiselenoate)、phosphoroanilothioate、phosphoraniladate、氨基磷酸酯(phosphoramidate)等。参见,例如laplanche等人nucleicacidsres.14:9081(1986);stec等人j.am.chem.soc.106:6077(1984),stein等人nucleicacidsres.16:3209(1988),zon等人anticancerdrugdesign6:539(1991);zon等人oligonucleotidesandanalogues:apracticalapproach,第87-108页(f.eckstein编著,oxforduniversitypress,oxfordengland(1991));stec等人,第5,151,510号美国专利;uhlmannandpeymanchemicalreviews90:543(1990)。在一些实施方案中,aso的主链结构不含有磷而含有肽键,例如在肽核酸(pna)中,或含有连接基团,包括氨基甲酸酯、酰胺以及直链烃基团和环状烃基团。在一些实施方案中,主链修饰是硫代磷酸酯键。在一些实施方案中,主链修饰是氨基磷酸酯键。
本文所述的任何aso可含有包含如天然存在的核苷酸中存在的核糖或脱氧核糖的糖部分,或含有包括吗啉环在内的经修饰的糖部分或糖类似物。经修饰的糖部分的非限制性实例包括2'取代,如2'-o-甲基(2'-o-me)、2'-o-甲氧乙基(2'moe)、2'-o-氨基乙基、2'f;n3’->p5’氨基磷酸酯、2'二甲基氨氧基乙氧基、2'二甲基氨基乙氧基乙氧基、2'-胍、2'-o-胍乙基、氨基甲酸酯修饰的糖和双环修饰的糖。在一些实施方案中,该糖部分修饰选自2′-o-me、2’f和2’moe。在一些实施方案中,该糖部分修饰是额外的桥键,如在锁定核酸(lna)中。在一些实施方案中,该糖类似物含有吗啉环,如磷二酰胺吗啉基(pmo)。在一些实施方案中,该糖部分包含呋喃核糖基或2’呋喃脱氧核糖基修饰。在一些实施方案中,该糖部分包含2’4’-约束的2’o-甲氧乙基(cmoe)修饰。在一些实施方案中,该糖部分包含cet2’,4’约束的2’o-乙基bna修饰。在一些实施方案中,该糖部分包含三环dna(tcdna)修饰。在一些实施方案中,该糖部分包含乙烯核酸(ena)修饰。在一些实施方案中,该糖部分包含mce修饰。修饰是本领域已知的并且描述在文献中,例如,jarver等人,2014,“achemicalviewofoligonucleotidesforexonskippingandrelateddrugapplications,”nucleicacidtherapeutics24(1):37-47,其通过引用并入本文用于此目的。
在一些实例中,aso的每个单体以相同的方式修饰,例如aso的主链的每个连接包含硫代磷酸酯键或每个核糖糖部分包含2’o-甲基修饰。存在于aso的每个单体组分上的此类修饰被称为“均一修饰”。在一些实例中,可能需要不同修饰的组合,例如,aso可包含磷二酰胺键和含有吗啉环(吗啉基)的糖部分的组合。aso的不同修饰的组合被称为“混合修饰”或“混合化学”。
在一些实施方案中,所述aso包含一个或多个主链修饰。在一些实施方案中,该aso包含一个或多个糖部分修饰。在一些实施方案中,该aso包含一个或多个主链修饰和一个或多个糖部分修饰。在一些实施方案中,该aso包含2’moe修饰和硫代磷酸酯主链。在一些实施方案中,该aso包含磷二酰胺吗啉基(pmo)。在一些实施方案中,该aso包含肽核酸(pna)。本文所述的任何aso或aso的任何组分(例如,核碱基、糖部分、主链)均可进行修饰,以便获得aso的所需性质或活性或减少aso的不需要的性质或活性。例如,aso或任何aso的一个或多个组分可被修饰为增强与前mrna转录物上靶序列的结合亲和力;减少与任何非靶序列的结合;减少被细胞核酸酶(即,核糖核酸酶h)的降解;改善aso向细胞中和/或向细胞的细胞核中的吸收;改变aso的药代动力学或药效学;以及调节aso的半衰期。
在一些实施方案中,所述aso由2'-o-(2-甲氧乙基)(moe)硫代磷酸酯修饰的核苷酸组成。由此类核苷酸组成的aso尤其适合于本文公开的方法;具有此类修饰的寡聚体已显示出具有显著增强的对核酸酶降解的抗性和增加的生物利用度,使得它们适合于例如在本文所述的一些实施方案中的口服递送。参见,例如,geary等人,jpharmacolexpther.2001;296(3):890-7;geary等人,jpharmacolexpther.2001;296(3):898-904。
本领域技术人员将知晓合成aso的方法。备选地或另外地,aso可从商业来源获得。
除非另有规定,否则单链核酸(例如,前mrna转录物、寡核苷酸、aso等)序列的左手端为5’端,并且单链或双链核酸序列的左手方向被称为5’方向。类似地,核酸序列(单链或双链)的右手端或右手方向为3’端或3’方向。通常,核酸中5’到参考点的区域或序列被称为“上游”,而核酸中3’到参考点的区域或序列被称为“下游”。通常,mrna的5’方向或5’端是启动或起始密码子所处的位置,而3’端或3’方向是终止密码子所处的位置。在一些方面,在核酸中参考点上游的核苷酸可以用负数加以标明,而在参考点下游的核苷酸则可以用正数加以标明。例如,参考点(例如,mrna中的外显子-外显子接合点)可标为“零”点,并且直接相邻且在该参考点上游的核苷酸标为“负一”,例如,“-1”,而直接相邻且在该参考点下游的核苷酸标为“正一”,例如,“+1”。
在其他实施方案中,所述aso互补于(且结合)在ric前mrna中保留内含子的5’剪接位点下游(在3’方向)的ric前mrna的靶向部分(例如,相对于5’剪接位点的以正数标明的方向)(图1)。在一些实施方案中,该aso互补于在相对于保留内含子的5’剪接位点的+6至+100区域内的ric前mrna的靶向部分。在一些实施方案中,该aso不互补于相对于5’剪接位点的+1至+5核苷酸(位于5’剪接位点下游的前五个核苷酸)。在一些实施方案中,该aso可互补于在相对于保留内含子的5’剪接位点的+6至+50之间核苷酸区域内的ric前mrna的靶向部分。在一些方面,该aso互补于在相对于保留内含子的5’剪接位点的+6至+90、+6至+80、+6至+70、+6至+60、+6至+50、+6至+40、+6至+30或+6至+20区域内的靶向部分。
在一些实施方案中,该aso互补于在ric前mrna中保留内含子的3’剪接位点上游(5’相对的)的ric前mrna的靶向部分(例如,在以负数标明的方向)(图1)。在一些实施方案中,该aso互补于在相对于保留内含子的3’剪接位点的-16至-100区域内的ric前mrna的靶向部分。在一些实施方案中,该aso不互补于相对于3’剪接位点的-1至-15核苷酸(位于3’剪接位点上游的前15个核苷酸)。在一些实施方案中,该aso互补于在相对于保留内含子的3’剪接位点的-16至-50区域内的ric前mrna的靶向部分。在一些方面,该aso互补于在相对于保留内含子的3’剪接位点的-16至-90、-16至-80、-16至-70、-16至-60、-16至-50、-16至-40或-16至-30区域内的靶向部分。
在实施方案中,ric前mrna的靶向部分在相对于保留内含子的5’剪接位点的+100至相对于保留内含子的3’剪接位点的-100的区域内。
在一些实施方案中,所述aso互补于在保留内含子的5’剪接位点侧翼(上游)的外显子内的ric前mrna的靶向部分(图1)。在一些实施方案中,该aso互补于在保留内含子的5’剪接位点侧翼的外显子中+2e至-4e区域内的ric前mrna的靶向部分。在一些实施方案中,该aso不互补于相对于保留内含子的5’剪接位点的-1e至-3e核苷酸。在一些实施方案中,该aso互补于在相对于保留内含子的5’剪接位点的-4e至-100e、-4e至-90e、-4e至-80e、-4e至-70e、-4e至-60e、-4e至-50e、-4至-40e、-4e至-30e或-4e至-20e区域内的ric前mrna的靶向部分。
在一些实施方案中,该aso互补于在保留内含子的3’剪接位点侧翼(下游)的外显子内的ric前mrna的靶向部分(图1)。在一些实施方案中,该aso互补于在保留内含子的3’剪接位点侧翼的外显子中+2e至-4e区域内的ric前mrna的靶向部分。在一些实施方案中,该aso不互补于相对于保留内含子的3’剪接位点的+1e核苷酸。在一些实施方案中,该aso互补于在相对于保留内含子的3’剪接位点的+2e至+100e、+2e至+90e、+2e至+80e、+2e至+70e、+2e至+60e、+2e至+50e、+2e至+40e、+2e至+30e或+2至+20e区域内的ric前mrna的靶向部分。该aso可以是适合于特异性结合和有效加强剪接的任何长度。在一些实施方案中,该aso由8至50个核碱基组成。例如,该aso的长度可以为8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、40、45或50个核碱基。在一些实施方案中,该aso由多于50个核碱基组成。在一些实施方案中,该aso的长度为8至50个核碱基、8至40个核碱基、8至35个核碱基、8至30个核碱基、8至25个核碱基、8至20个核碱基、8至15个核碱基、9至50个核碱基、9至40个核碱基、9至35个核碱基、9至30个核碱基、9至25个核碱基、9至20个核碱基、9至15个核碱基、10至50个核碱基、10至40个核碱基、10至35个核碱基、10至30个核碱基、10至25个核碱基、10至20个核碱基、10至15个核碱基、11至50个核碱基、11至40个核碱基、11至35个核碱基、11至30个核碱基、11至25个核碱基、11至20个核碱基、11至15个核碱基、12至50个核碱基、12至40个核碱基、12至35个核碱基、12至30个核碱基、12至25个核碱基、12至20个核碱基、12至15个核碱基、13至50个核碱基、13至40个核碱基、13至35个核碱基、13至30个核碱基、13至25个核碱基、13至20个核碱基、14至50个核碱基、14至40个核碱基、14至35个核碱基、14至30个核碱基、14至25个核碱基、14至20个核碱基、15至50个核碱基、15至40个核碱基、15至35个核碱基、15至30个核碱基、15至25个核碱基、15至20个核碱基、20至50个核碱基、20至40个核碱基、20至35个核碱基、20至30个核碱基、20至25个核碱基、25至50个核碱基、25至40个核碱基、25至35个核碱基或25至30个核碱基。在一些实施方案中,该aso的长度为18个核苷酸。在一些实施方案中,该aso的长度为15个核苷酸。在一些实施方案中,该aso的长度为25个核苷酸。
在一些实施方案中,使用具有不同化学但与ric前mrna的相同靶向部分互补的两个或更多个aso。在一些实施方案中,使用与ric前mrna的不同靶向部分互补的两个或更多个aso。
在实施方案中,本发明的反义寡核苷酸经化学连接至一个或多个部分或缀合物,例如,增强该寡核苷酸的活性或细胞吸收的靶向部分或其他缀合物。此类部分包括但不限于脂质部分,例如,胆固醇部分、胆固醇基部分、脂肪链,例如,十二烷二醇或十一烷基残基、多胺或聚乙二醇链或金刚烷乙酸。在公开的文献中已描述了包含亲脂性部分的寡核苷酸和制备方法。在实施方案中,该反义寡核苷酸与某部分缀合,该部分包括但不限于无碱基核苷酸、聚醚、多胺、聚酰胺、肽、碳水化合物,例如,n-乙酰半乳糖胺(galnac)、n-ac-葡糖胺(glunac)或甘露糖(例如,甘露糖-6-磷酸酯)、脂质或聚烃化合物。如本领域理解的和文献中描述的,例如可使用连接体将缀合物与构成反义寡核苷酸的任何核苷酸中的一个或多个在糖、碱基或磷酸基团上若干位置的任何一处连接。连接体可包括二价或三价分支连接体。在实施方案中,该缀合物附接至该反义寡核苷酸的3’端。制备寡核苷酸缀合物的方法描述在例如第8,450,467号美国专利,“carbohydrateconjugatesasdeliveryagentsforoligonucleotides”中,其通过引用并入本文。
在一些实施方案中,被aso靶向的核酸是在细胞如真核细胞中表达的ric前mrna。在一些实施方案中,术语“细胞”可指细胞群体。在一些实施方案中,该细胞在受试者中。在一些实施方案中,该细胞从受试者中分离。在一些实施方案中,该细胞是离体的。在一些实施方案中,该细胞是病况或疾病有关的细胞或细胞系。在一些实施方案中,该细胞在体外(例如,在细胞培养物中)。
药物组合物
包含所述组合物的反义寡核苷酸且用于任何所述方法的药物组合物或制剂可根据制药工业中公知的和公开文献中描述的常规技术进行制备。在实施方案中,用于治疗受试者的药物组合物或制剂包含有效量的上述任何反义寡聚体,或其药学上可接受的盐、溶剂化物、水合物或酯,以及药学上可接受的稀释剂。药物制剂的反义寡聚体可进一步包含药学上可接受的赋形剂、稀释剂或载体。
药学上可接受的盐适用于与人和低等动物的组织相接触而没有不当的毒性、刺激、变态反应等,并且对应合理的受益/风险比。参见,例如,s.m.berge等人,j.pharmaceuticalsciences,66:1-19(1977),其通过引用并入本文用于此目的。所述盐可以在化合物的最终分离和纯化期间原位制备,或通过使游离碱官能与合适的有机酸反应予以单独制备。药学上可接受的、无毒的酸加成盐的实例是氨基基团与无机酸如盐酸、氢溴酸、磷酸、硫酸和高氯酸或与有机酸如乙酸、草酸、马来酸、酒石酸、柠檬酸、琥珀酸或丙二酸所形成的盐,或通过利用其他记录的方法如离子交换形成的盐。其他药学上可接受的盐包括己二酸盐、藻酸盐、抗坏血酸盐、天冬氨酸盐、苯磺酸盐、苯甲酸盐、硫酸氢盐、硼酸盐、丁酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、环戊烷丙酸盐、二葡糖酸盐、十二烷基硫酸盐、乙磺酸盐、甲酸盐、富马酸盐、葡庚糖酸盐、甘油磷酸盐、葡糖酸盐、半硫酸盐、庚酸盐、己酸盐、氢碘酸盐、2-羟基-乙磺酸盐、乳糖酸盐、乳酸盐、月桂酸盐、月桂基硫酸盐、苹果酸盐、马来酸盐、丙二酸盐、甲磺酸盐、2-萘磺酸盐、烟酸盐、硝酸盐、油酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、果胶酸盐、过硫酸盐、3-苯基丙酸盐、磷酸盐、苦味酸盐、新戊酸盐、丙酸盐、硬脂酸盐、琥珀酸盐、硫酸盐、酒石酸盐、硫氰酸盐、对甲苯磺酸盐、十一酸盐、戊酸盐等。代表性的碱金属或碱土金属盐包括钠、锂、钾、钙、镁盐等。视情况而定,另外的药学上可接受的盐包括利用抗衡离子如卤离子、氢氧根、羧酸根、硫酸根、磷酸根、硝酸根、低级烷基磺酸根和芳基磺酸根与铵、季铵和胺阳离子形成的无毒盐。
在实施方案中,将组合物配制为许多可能剂型中的任何一种,例如但不限于片剂、胶囊、凝胶胶囊、液体糖浆、软凝胶、栓剂和灌肠剂。在实施方案中,将组合物在水性、非水性或混合介质中配制为悬浮液。水性悬浮液可进一步含有增加该悬浮液的粘度的物质,包括例如羧甲基纤维素钠、山梨糖醇和/或葡聚糖。该悬浮液还可以含有稳定剂。在实施方案中,本发明的药物制剂或组合物包括但不限于溶液、乳液、微乳液、泡沫或含脂质体的制剂(例如,阳离子型或非阳离子型脂质体)。
本发明的药物组合物或制剂可包含视情况而定的且本领域技术人员公知的或公开文献中描述的一种或多种渗透促进剂、载体、赋形剂或其他活性或非活性成分。在实施方案中,脂质体还包括空间稳定的脂质体,例如,包含一种或多种特化脂质的脂质体。这些特化脂质导致具有延长的循环寿命的脂质体。在实施方案中,空间稳定的脂质体包含一种或多种糖酯,或用一种或多种亲水性聚合物如聚乙二醇(peg)部分来衍生。在实施方案中,所述药物制剂或组合物中包含表面活性剂。在药物产品、制剂和乳液中使用表面活性剂是本领域公知的。在实施方案中,本发明采用渗透促进剂来实现反义寡核苷酸的有效递送,例如,以帮助跨细胞膜的扩散和/或加强亲脂性药物的渗透性。在实施方案中,该渗透促进剂是表面活性剂、脂肪酸、胆汁盐、螯合剂或非螯合非表面活性剂。
在实施方案中,所述药物制剂包含多种反义寡核苷酸。在实施方案中,该反义寡核苷酸与另一种药物或治疗剂联合施用。在实施方案中,该反义寡核苷酸通过本领域已知的任何方法与能够促进该反义寡核苷酸跨越血脑屏障渗透的一种或多种药剂一起施用。例如,在第6,632,427号美国专利,"adenoviral-vector-mediatedgenetransferintomedullarymotorneurons"中描述了通过将腺病毒载体施用于肌肉组织中的运动神经元来递送药剂,其通过引用并入本文。例如,在第6,756,523号美国专利,"adenovirusvectorsforthetransferofforeigngenesintocellsofthecentralnervoussystemparticularlyinbrain"中描述了将载体直接递送至脑,例如,纹状体、丘脑、海马体或黑质体,其通过引用并入本文。
在实施方案中,所述反义寡核苷酸与提供所需药物或药效学性质的药剂连接或缀合。在实施方案中,该反义寡核苷酸与本领域已知的物质例如转铁蛋白受体的抗体偶联以促进跨越血脑屏障的渗透或转运。在实施方案中,该反义寡核苷酸与病毒载体连接,例如,以使该反义化合物更有效或增加跨越血脑屏障的转运。在实施方案中,通过输注糖,例如内消旋赤藓醇、木糖醇、d(+)半乳糖、d(+)乳糖、d(+)木糖、卫矛醇、肌醇、l(-)果糖、d(-)甘露醇、d(+)葡萄糖、d(+)阿拉伯糖、d(-)阿拉伯糖、纤维二糖、d(+)麦芽糖、d(+)棉子糖、l(+)鼠李糖、d(+)蜜二糖、d(-)核糖、侧金盏花醇、d(+)阿拉伯糖醇、l(-)阿拉伯糖醇、d(+)岩藻糖、l(-)岩藻糖、d(-)来苏糖、l(+)来苏糖和l(-)来苏糖,或氨基酸,例如谷氨酰胺、赖氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酸、甘氨酸、组氨酸、亮氨酸、甲硫氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、酪氨酸、缬氨酸和牛磺酸,来帮助渗透性血脑屏障破坏。用于加强血脑屏障渗透的方法和材料描述在例如第4,866,042号美国专利,"methodforthedeliveryofgeneticmaterialacrossthebloodbrainbarrier",第6,294,520号美国专利,"materialforpassagethroughtheblood-brainbarrier",以及第6,936,589号美国专利,"parenteraldeliverysystems"中,其每一个均通过引用并入本文。
在实施方案中,本发明的反义寡核苷酸经化学连接至一个或多个部分或缀合物,例如,增强该寡核苷酸的活性或细胞吸收的靶向部分或其他缀合物。此类部分包括但不限于脂质部分,例如,胆固醇部分、胆固醇基部分、脂肪链,例如,十二烷二醇或十一烷基残基、多胺或聚乙二醇链或金刚烷乙酸。在公开的文献中已描述了包含亲脂性部分的寡核苷酸和制备方法。在实施方案中,该反义寡核苷酸与某部分缀合,该部分包括但不限于无碱基核苷酸、聚醚、多胺、聚酰胺、肽、碳水化合物,例如,n-乙酰半乳糖胺(galnac)、n-ac-葡糖胺(glunac)或甘露糖(例如,甘露糖-6-磷酸酯)、脂质或聚烃化合物。如本领域理解的和文献中描述的,例如可使用连接体将缀合物与构成反义寡核苷酸的任何核苷酸中的一个或多个在糖、碱基或磷酸基团上若干位置的任何一处连接。连接体可包括二价或三价支链连接体。在实施方案中,该缀合物附接至该反义寡核苷酸的3’端。制备寡核苷酸缀合物的方法描述在例如第8,450,467号美国专利,“carbohydrateconjugatesasdeliveryagentsforoligonucleotides”中,其通过引用并入本文。
疾病和病症
与由包含至少一个内含子(例如,1、2、3、4、5、6、7、8、9、10个或更多个内含子)的前mrna编码的蛋白质或功能rna的产生量或活性降低相关的任何病况,例如,疾病或病症,可通过本文提供的方法和组合物予以治疗。待治疗的疾病或病症可以是单倍性不足的结果,其中基因的一个等位基因编码功能(野生型)蛋白质而该基因的一个等位基因被突变且编码非功能性蛋白质或具有降低功能/部分功能的蛋白质。其他疾病或病症可由半合子缺失引起,其中基因的一个等位基因丢失且由该基因的另一个等位基因所产生的蛋白质的量不足。其他疾病或病症可能由亚等位基因突变引起,其中编码蛋白质的基因被突变,导致产生了具有部分功能的蛋白质。
在一些实施方案中,本文所述方法用来增加功能蛋白质的产生量。如本文所用,术语“功能性”是指消除疾病的任何一种或多种症状所需的蛋白质的活性的量或功能。在一些实施方案中,所述方法用来增加部分功能性蛋白质或rna的产生量。如本文所用,术语“部分功能性”是指低于消除或预防疾病的任何一种或多种症状所需的活性量或功能的蛋白质或rna的任何活性量或功能。在一些实施方案中,部分功能性蛋白质或rna将具有比完全功能性蛋白质或rna低至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少75%、至少80%、85%、至少90%或至少95%的活性。
在实施方案中,所述方法是一种增加具有编码靶蛋白或功能rna的ric前mrna的受试者细胞表达该靶蛋白或功能rna的方法,其中该受试者具有由该靶蛋白或功能rna的活性的量缺陷而引起的病况,并且其中该靶蛋白或功能rna的量缺陷由该靶蛋白或功能rna的单倍性不足而引起。在这样的实施方案中,受试者具有编码功能性靶蛋白或功能性的功能rna的第一等位基因,和不产生该靶蛋白或功能rna的第二等位基因。在另一个这样的实施方案中,受试者具有编码功能性靶蛋白或功能性的功能rna的第一等位基因,和编码非功能性靶蛋白或非功能性的功能rna的第二等位基因。在这些实施方案的任何一个中,所述反义寡聚体与由第一等位基因(编码功能性靶蛋白)转录的ric前mrna的靶向部分结合,从而诱导保留内含子从该ric前mrna中组成性剪接,并引起编码靶蛋白或功能rna的mrna的水平增加,以及在受试者细胞中该靶蛋白或功能rna的表达增加。
在相关的实施方案中,所述方法是一种增加具有编码靶蛋白或功能rna的ric前mrna的受试者细胞表达该靶蛋白或功能rna的方法,其中该受试者具有由常染色体隐性失常引起的病况,该常染色体隐性失常由该靶蛋白或功能rna的量或功能缺陷而导致。在这些实施方案中,受试者具有:
a.第一突变体等位基因,由其
i)产生该靶蛋白或功能rna的水平与由野生型等位基因产生相比降低,
ii)产生与对应的野生型蛋白相比功能降低的形式的该靶蛋白或功能rna,或
iii)不产生该靶蛋白或功能rna;以及
b.第二突变体等位基因,由其
i)产生该靶蛋白或功能rna的水平与从野生型等位基因产生相比降低,
ii)产生与对应的野生型蛋白相比功能降低的形式的该靶蛋白或功能rna,或
iii)不产生该靶蛋白或功能rna,并且
其中所述ric前mrna由第一等位基因和/或第二等位基因转录。在这些实施方案中,所述反义寡聚体与由第一等位基因或第二等位基因所转录的ric前mrna的靶向部分结合,从而诱导保留内含子从该ric前mrna中的组成性剪接,并引起编码靶蛋白或功能rna的mrna的水平增加,以及在受试者细胞中靶蛋白或功能rna的表达增加。在这些实施方案中,由保留内含子从ric前mrna中组成性剪接导致表达水平增加的靶蛋白或功能rna为与对应的野生型蛋白相比功能降低的形式(部分功能性),或与对应的野生型蛋白相比具有完全功能的形式(完全功能性)。
在实施方案中,如本文别处所述,当与在对照细胞(例如,未用反义寡聚体处理的细胞或用不与ric前mrna的靶向部分结合的反义寡聚体处理的细胞)中产生的编码靶蛋白的mrna、该靶蛋白或功能rna的量相比时,编码该靶蛋白的mrna、该靶蛋白或该功能rna的水平增加至1.1至10倍。
在实施方案中,由靶蛋白的量或活性缺陷或功能rna的量或活性缺陷引起的病况不是由aso所靶向的保留内含子的其它或异常剪接所引起的病况。在实施方案中,由靶蛋白的量或活性缺陷或功能rna的量或活性缺陷引起的病况不是由在编码该靶蛋白或功能rna的ric前mrna中的任何保留内含子的其它或异常剪接所引起的病况。
表1提供了疾病和靶基因的实例,所述靶基因与可利用本文提供的方法和组合物治疗的每种疾病相关。
表1
在一些实施方案中,编码引起疾病的蛋白质的前mrna转录物被本文所述的aso所靶向。在一些实施方案中,编码不引起疾病的蛋白质的前mrna转录物被所述aso所靶向。例如,作为特定途径中第一蛋白质突变或缺陷的结果的疾病可通过靶向编码第二蛋白质的前mrna,从而增加该第二蛋白质的产生量来改善。在一些实施方案中,第二蛋白质的功能能够补偿第一蛋白质的突变或缺陷。
可将本文提供的任何组合物施用于个体。“个体”可与“受试者”或“患者”互换使用。个体可以是哺乳动物,例如人或动物,如非人灵长类、啮齿动物、兔、大鼠、小鼠、马、驴、山羊、猫、狗、牛、猪或绵羊。在一些实施方案中,个体是人。在其他的实施方案中,个体可以是另一种真核生物,如植物。在一些实施方案中,将本文提供的组合物施用于离体细胞。
在一些实施方案中,将本文提供的组合物作为治疗疾病或病症的方法施用于个体。在一些实施方案中,该个体患有遗传病,如本文所述的任何疾病。在一些实施方案中,该个体处于患有疾病如本文所述的任何疾病的风险中。在一些实施方案中,该个体处于患有由蛋白质的量不足或蛋白质的活性不足引起的疾病或病症的增加的风险中。如果个体处于患有蛋白质的量不足或蛋白质的活性不足引起的疾病或病症的“增加的风险”中,则该方法包括预防性或防范性处理。例如,个体可能由于该疾病的家族史而处于患有这样的疾病或病症的增加的风险中。通常,处于患有这样的疾病或病症的增加的风险中的个体受益于防范性处理(例如,通过预防或延迟该疾病或病症的发作或进展)。
表2提供了用于通过靶向由hbb基因转录的ric前mrna的区域来增加由hbb基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表2.靶向hbb基因的aso的列表
表3提供了用于通过靶向由prpf31基因转录的ric前mrna的区域来增加由prpf31基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表3.靶向prpf31基因的aso的列表
表4提供了用于通过靶向由adamts13基因转录的ric前mrna的区域来增加由adamts13基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表4.靶向adamts13基因的aso的列表
表5提供了用于通过靶向由tsc1基因转录的ric前mrna的区域来增加由tsc1基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表5.靶向tsc1基因的aso的列表
表6提供了用于通过靶向由impdh1基因转录的ric前mrna的区域来增加由impdh1基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表6.靶向impdh1基因的aso的列表
表7提供了用于通过靶向由pkd1基因转录的ric前mrna的区域来增加由pkd1基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表7.靶向pkd1基因的aso的列表
表8提供了用于通过靶向由ikbkap基因转录的ric前mrna的区域来增加由ikbkap基因编码的蛋白质的产生量的aso的序列的非限制性列表。
表8.靶向ikbkap基因的aso的列表
鉴定保留内含子的方法
在本公开内容的范围内还包括鉴定(确定)当相邻(上游或下游)内含子从细胞中的前mrna中剪接掉时前mrna转录物中的保留内含子的方法。在一个实例中,可通过以下方法测量外显子剪接和连接以及每个内含子从靶基因中去除的程度。本领域技术人员将会理解,可以使用任何方法来确定内含子是否相对于从前mrna转录物中剪接掉的相邻内含子而保留在前mrna转录物中,以及靶内含子是否相对于在由相同基因编码的前mrna内的一个或多个其他内含子而在较大程度上得以保留。
i.筛选保留内含子
可利用从细胞或组织(例如,疾病相关细胞)中分离出的细胞核rna进行内含子保留的第一轮筛选,并通过逆转录酶-pcr(rt-pcr)进行分析,例如,以研究由靶基因编码的前rna。靶基因可以是含有至少一个内含子且编码与疾病或病症相关的或怀疑相关的或引起疾病或病症的蛋白质或功能rna的任何基因。对于rt-pcr分析,通过设计一系列引物对评估由基因编码的前mrna中保留的每一个内含子,其中该引物对中的一个引物针对该靶前mrna的内含子的区域是特异性的,并且该引物对中的另一个引物针对外显子的区域是特异性的,该外显子区域为该内含子上游或下游的两个外显子(图3)。在一些实施方案中,上游或正向引物可与内含子(例如,图3中外显子1与2之间的内含子)内的区域互补和杂交;而下游或反向引物可与位于与所评估的内含子相距两个外显子的外显子内(例如在如图3所示的外显子3内)的区域互补和杂交。或者,上游或正向引物可与外显子内(例如,图3中在外显子2中)的区域互补和杂交;而下游或反向引物可与距正向引物两个外显子的内含子内(例如在如图3所示的外显子3与4之间的内含子内)的区域互补和杂交。可针对每一个由基因编码的内含子来重复引物对的设计。
采用每一个引物对进行rt-pcr后,通过本领域已知的任何方法分析rt-pcr产物,例如,在琼脂糖凝胶中分离和可视化。如果存在靶内含子,则预期的rt-pcr产物的近似大小可根据基因和/或前mrna的核酸序列来估算。rt-pcr分析不存在产物表明,靶内含子不存在且已从前mrna中去除/剪接,因此在所测试的条件下其并非保留内含子。rt-pcr反应存在约等于估算的产物大小的产物表明,靶内含子存在于前mrna中且在测试条件下未从前mrna中去除/剪接,此类内含子被称为“保留内含子”。
在需要对许多前rna或在整个转录物组水平上进行分析的实例中,可通过rna-seq或任何其他高通量转录分析方法来分析内含子保留的筛选。利用合适的深度测序读数的定位和统计方法进行rna-seq分析,来测定整个转录物组中的内含子保留事件。
ii.内含子保留事件的确认
可进行前mrna内的内含子的第二轮筛选以利用诸如rt-pcr等方法来确认内含子保留事件。可再次评估在上述第一轮筛选时被鉴定为保留内含子的每一个内含子。对于rt-pcr分析,通过设计引物对来评估由基因编码的前mrna中保留的每一个保留内含子,其中该引物对中的一个引物针对该靶前mrna的内含子的区域是特异性的,并且该引物对中的另一个引物针对外显子的区域是特异性的,该外显子区域为该内含子上游或下游的三个、四个或五个外显子(图4)。在图4所示的示意图中,待评估的保留内含子位于外显子1与2之间。上游或正向引物针对一个区域是特异性的,并在该保留内含子内杂交,而下游或反向引物被设计为与外显子4、外显子5和外显子6中的区域杂交,这些外显子分别与该保留内含子相距3、4和5个外显子。利用所述正向引物和每一个所述反向引物进行rt-pcr反应。
rt-pcr后,通过本领域已知的任何方法分析rt-pcr产物,例如,在琼脂糖凝胶中分离和可视化。根据来自每一个反应的rt-pcr产物的分子大小,可以确定除所测试的内含子(以上鉴定的保留内含子)之外每一个内含子(例如,外显子2与3、3与4以及4与5之间的内含子)是否也得到保留。当已去除/剪接一个或多个相邻内含子时被发现保留的保留内含子可被称为“低效剪接内含子”。
iii.测定内含子剪接效率
可以进一步评估相对于相同前mrna中被去除/剪接的其他内含子被鉴定为持久内含子或低效剪接内含子的由靶基因编码的前mrna中的任何内含子,以测定内含子保留的比例或效率。
可通过进行测定如核糖核酸酶保护测定来评估内含子以测定内含子保留的效率(图5)。设计一对rna探针(例如,放射性标记的rna探针),其中每一个探针对跨越保留内含子和相邻外显子的末端的区域是特异性的。例如,rna探针被设计为与跨越保留内含子的5’端和保留内含子上游的外显子的3’端的区域杂交;而第二rna探针被设计为与跨越保留内含子的3’端和保留内含子下游的外显子的5’端的区域杂交。在一些实施方案中,与该内含子杂交的探针部分的长度为至少100个核苷酸,而与该外显子杂交的探针部分的长度为至少50个核苷酸(图5)。在探针与前mrna的区域杂交形成双链rna的区域的条件下,将从疾病相关细胞、组织或细胞系中提取的核rna与rna探针对一起温育。前mrna和rna探针的混合物用降解单链rna的核糖核酸酶如核糖核酸酶a和/或核糖核酸酶t1进行消化。双链rna不被降解。
通过本领域已知的任何方法分析核糖核酸酶消化反应,例如,在琼脂糖凝胶中分离和可视化。对应于rna探针全长(例如,150个核苷酸)的rna分子的量指示前mrna中存在的保留内含子的量。对应于消化的rna探针的rna分子(例如,长度为约50个核苷酸的rna分子)的量代表当与rna探针杂交的内含子不存在于前mrna中时(例如,被剪接掉)剪接的rna的量。内含子保留(全长rna探针的量,例如,100个核苷酸的rna分子)与剪接rna(降解的rna探针的量,例如,50个核苷酸的rna分子)的比例指示内含子的剪接效率。相对于相同前mrna的其他内含子的比例为最高的前mrna的内含子指示该内含子是由靶基因编码的前mrna的最低效剪接内含子或最高保留内含子。
鉴定增强剪接的aso的方法
本发明的范围内还包括用于鉴定(确定)增强靶前mrna(特别是靶内含子处)的剪接的aso的方法。可筛选与前mrna的靶区域内的不同核苷酸特异性杂交的aso以鉴定(确定)增加靶内含子的剪接速度和/或程度的aso。在一些实施方案中,该aso可阻断或干扰剪接阻抑物/沉默基因的结合位点。可利用本领域已知的任何方法来鉴定(确定)当与内含子的靶区域杂交时导致所需效果(例如,提高的剪接、蛋白质或功能rna产生)的aso。这些方法还可用于鉴定通过与位于保留内含子侧翼的外显子中或非保留内含子中的靶向区域结合而增强保留内含子的剪接的aso。以下提供了可采用的方法的实例。
被称为aso“步移”的一轮筛选可采用被设计为与前mrna的靶区域杂交的aso来进行。例如,aso步移中所用的aso可从保留内含子的5’剪接位点上游约100个核苷酸(例如,位于靶/保留内含子上游的外显子序列的一部分)至该靶/保留内含子的5’剪接位点下游约100个核苷酸,和/或从该保留内含子的3’剪接位点上游约100个核苷酸至该靶/保留内含子的3’剪接位点下游约100个核苷酸(例如,位于该靶/保留内含子下游的外显子序列的一部分)以每5个核苷酸进行平铺。例如,长度为15个核苷酸的第一aso可设计为与相对于靶/保留内含子的5’剪接位点的+6至+20核苷酸特异性杂交。将第二aso设计为与相对于靶/保留内含子的5’剪接位点的+11至+25核苷酸特异性杂交。将aso设计为跨越前mrna的靶区域。在实施方案中,aso可更紧密地平铺,例如,每1个、2个、3个或4个核苷酸。此外,aso可从5’剪接位点下游的100个核苷酸至3’剪接位点上游的100个核苷酸进行平铺。
例如,通过转染将一个或多个aso或对照aso(具有错义序列——预期不与靶区域杂交的序列——的aso)递送至表达靶前mrna(例如,本文别处所述的ric前mrna)的疾病相关细胞系中。如本文所述(参见“内含子保留事件的鉴定”),可通过本领域已知的任何方法,例如通过使用跨越剪接点的引物的逆转录酶(rt)-pcr,评估每一个aso的剪接诱导效果。与在对照aso处理的细胞中相比,在aso处理的细胞中使用跨越剪接点的引物产生的rt-pcr产物的减少或不存在表明靶内含子的剪接已得到加强。在一些实施方案中,可利用本文所述的aso来提高剪接效率、剪接的与未剪接的前mrna之比、剪接速度或剪接程度。还可以评估由靶前mrna编码的蛋白质或功能rna的量以确定每一个aso是否均获得所需效果(例如,增加的蛋白质产生量)。可以使用本领域已知用于评估和/或量化蛋白质产生量的任何方法,如蛋白质印迹法、流式细胞术、免疫荧光显微术和elisa。
可利用被设计为与前mrna的靶区域杂交的aso来进行被称为aso“微步移”的第二轮筛选。aso微步移中所用的aso以每隔1个核苷酸进行平铺,以进一步精修当与aso杂交时导致剪接增强的前mrna的核苷酸序列。
借助于aso“微步移”,包括以1-nt步长间隔的aso以及更长的aso(通常为18-25nt),更详细地探索了由促进靶内含子的剪接的aso所限定的区域。
如上对于aso步移所述,通过将一个或多个aso或对照aso(具有错乱(scrambled)序列——预期不与靶区域杂交的序列——的aso)例如通过转染递送至表达靶前mrna的疾病相关细胞系中来进行aso微步移。如本文所述(参见“内含子保留事件的鉴定”),可通过本领域已知的任何方法,例如通过采用跨越剪接点的引物的逆转录酶(rt)-pcr,评估每一个aso的剪接诱导效果。与在对照aso处理的细胞中相比,在aso处理的细胞中采用跨越剪接点的引物所产生的rt-pcr产物的减少或不存在表明靶内含子的剪接已经得到增强。在一些实施方案中,可利用本文所述的aso来提高剪接效率、已剪接的与未剪接的前mrna之比、剪接速度或剪接程度。还可以评估由靶前mrna编码的蛋白质或功能rna的量以确定每一个aso是否均达到所需效果(例如,增加的蛋白质产生量)。可以使用本领域已知用于评估和/或量化蛋白质产生量的任何方法,如蛋白质印迹法、流式细胞术、免疫荧光显微术和elisa。
当与前mrna的区域杂交时导致增强的剪接和增加的蛋白质产生量的aso可利用动物模型,例如已敲入全长人基因的转基因小鼠模型,或在疾病的人源化小鼠模型中进行体内测试。合适的aso施用途径可根据需要递送aso的疾病和/或细胞类型而变化。例如,可通过玻璃体内注射、鞘内注射、腹膜内注射、皮下注射或静脉内注射来施用aso。施用后,可评估模型动物的细胞、组织和/或器官,以通过例如由本领域已知和本文所述的方法评价剪接(效率、速度、程度)和蛋白质产生量来确定aso治疗的效果。动物模型还可以是疾病或疾病严重程度的任何表型或行为指示。
实施例
本发明将通过以下实施例更具体地加以说明。然而,应当理解,本发明不以任何方式由这些实施例来限制。
实施例1:内含子保留事件是基因固有的,并且是非生产性的
利用本文所述方法对prpf31(11型视网膜色素变性)和rb1(视网膜母细胞瘤)基因中的内含子保留事件进行第一轮筛选(图3)。简而言之,从hela(人上皮宫颈腺癌)和293t(人胚肾上皮)细胞的细胞核部分以及arpe-19(人视网膜)细胞的细胞核和细胞质部分中分离出rna提取物。采用来自每一个所述细胞类型的rna提取物进行逆转录酶pcr(rt-pcr)。简而言之,采用寡核苷酸dt进行cdna合成以仅生成聚arna(完全转录的rna)的dna拷贝,并进行pcr以评估prpf31和rb1转录物中的内含子保留。该pcr产物在1.5%溴化乙锭染色的琼脂糖凝胶上分离(图6a-图6d)。结果显示在测试的三种细胞系中的每一个的细胞核中两个基因(prpf31和rb1)的若干内含子保留事件(由黑色星号标记)(图6a-图6d)。
表9和表10列出了在分别针对prpf31和rb1测试的三种细胞系中发生的所有内含子保留事件。在所有三种细胞系中发生的事件(存在或不存在内含子保留)用星号指示。所述表格显示在这三种细胞系间存在非常高的一致性,这表明内含子保留事件是基因固有的,并且不受不同细胞环境的影响。
为了讨论这些事件是否为非生产性的(即能够导致蛋白质产生),采用arpe-19细胞的细胞质部分进行rt-pcr(图6e)。结果显示,大多数所观察到的内含子保留事件不存在于arpe-19细胞的细胞质中(图6e,星号标记条带应在的位置),正如预期的,表明内含子保留事件导致转录物保留在细胞核中或在细胞质中被无义介导的mrna分解所降解,并因此是非生产性的转录物。
表9:prpf31基因中内含子保留事件的结果的总结。“是”指示存在内含子保留;“否”指示不存在内含子保留;“?”指示无法作出结论的结果。在三种细胞系之间存在一致性的情况以星号标记。
表10.rb1基因中内含子保留事件的结果的总结。“是”指示存在内含子保留;“否”指示不存在内含子保留。在三种细胞系之间存在一致性的情况以星号标记。
实施例2:内含子保留事件的确认
使用本文所述方法对prpf31(11型视网膜色素变性)和rb1(视网膜母细胞瘤)基因中的内含子保留事件进行第二轮筛选(图4)。简而言之,如实施例1中所述采用来自arpe-19(人视网膜)细胞的细胞核rna提取物进行逆转录酶pcr(rt-pcr)。在该实施例中,在其中从前mrna中剪接掉(去除)超过一个内含子的情况下评估内含子保留。结果显示与图6a-图6d的结果相比,两个基因(prpf31和rb1)的内含子保留事件(用黑色星号标记)均较少(图7a-图7b),减少了候选内含子保留事件的数目。
实施例3:经由内含子区域的诱变或aso靶向而提高的剪接效率增加了基因表达
我们旨在提高与β地中海贫血有关的hbb(人β珠蛋白)基因的两个内含子中每一个的剪接效率,并评估这是否会导致转录物水平增加。整个hbb开放读框被克隆在小基因报道基因中。将突变引入两个内含子的5’和3’剪接位点,以便将它们带入完全共有序列。图8a示出了hbb基因和在剪接位点处引入的突变的示意图。使用fugene转染试剂将在每个剪接位点处携带突变以及这些突变的组合的小基因报道基因独立地转染至hek293(人胚肾上皮)细胞中24小时。放射性rt-pcr结果显示,仅改善内含子1(ivs1)的5’剪接位点的突变增加了hbb转录物水平(图8b)。对应于突变小基因的hbbpcr产物的条带强度的量化相对于gfp的条带强度进行归一化,并相对于野生型hbb进行绘图。这些柱条表明,当内含子1的剪接效率提高时,hbb的表达水平增加超过2倍(图8c)。我们先前已观察到,hbb内含子1是低效剪接的并且是该基因中的限速内含子(数据未示出)。我们在此证明了通过提高低效剪接内含子的剪接效率可以获得基因表达的显著增加。
目的是确定我们是否还可以通过利用aso提高hbb内含子1的剪接效率来获得hbb-报道基因(小基因)表达的增加。为此,生成18聚体2′-o-measo以靶向在位置+7处开始的内含子1,以及生成两个18聚体pmo-aso以靶向分别在相对于5’剪接点的位置+6和+7处开始的内含子1(图9a;表2,分别为seqidno:104和105)。首先利用fugene转染试剂用野生型hbb小基因报道基因和gfp(作为转染对照)共转染hek293细胞。四个小时后,利用rnaimax(rim)(invitrogen)或endoporter(ep)(genetools)递送试剂,对细胞进行未转染、模拟转染或用每一种靶向aso或非靶向aso对照独立地进行转染。使用如图9b所示浓度渐增的aso进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,具有两种化学的+7靶向aso增加了hbb转录物水平(图9b)。对于+6pmo-aso获得了类似的结果(数据未示出)。将对应于来自靶向-aso转染细胞的hbbpcr产物的条带强度相对于gfp进行归一化,并相对于来自模拟处理的细胞的归一化hbbpcr产物进行绘图。该分析的结果表明,两种靶向aso(+6和+7)使hbb转录物水平增加了将近50%(图9c)。这些结果表明,利用aso提高hbb基因中限速内含子的剪接效率导致基因表达增加。
实施例4:经由靶向内含子区域的aso而提高的剪接效率增加了蛋白质产生量
为了检测用+72′-o-measo靶向hbb内含子1时蛋白质产生量的增加,我们生成了由上游侧翼为gfp开放读框且下游侧翼为编码t7标签的序列的hbb小基因构成的报道基因构建体(图10a)。将该报道基因整合在模拟内源基因的u2os细胞的基因组中。模拟转染或用+72′-o-measo转染表达gfp-hbb-t7报道基因的u2os细胞,并通过蛋白质印迹法分析蛋白质提取物。简而言之,来自两个独立生物复制品的蛋白质提取物在4-20%sds-聚丙烯酰胺凝胶上运行,转移到硝酸纤维膜。为了证明蛋白质产生量的增加,采用抗gfp抗体来检测来自gfp-hbb-t7报道基因的蛋白质产物,并采用抗β微管蛋白抗体来检测作为加样对照的β微管蛋白。图10b示出了蛋白质印迹结果,其表明在用+72′-o-measo处理时增加了gfp-hbb-t7蛋白质(底部条带)。将对应于来自靶向-aso转染细胞的gfp-hbb-t7蛋白质的条带强度相对于内源性β微管蛋白进行归一化,并相对于来自模拟处理的细胞的归一化gfp-hbb-t7蛋白质条带进行绘图。
该分析的结果表明,靶向aso(+7)使gfp-hbb-t7蛋白水平增加超过2.5倍(图10c)。这些结果证实,通过使用靶向限速内含子的5’剪接位点下游区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所示。
实施例5:利用新一代测序通过rnaseq来鉴定adamts13转录物中的内含子保留事件
我们使用新一代测序进行全转录物组鸟枪法测序,以概略显示由adamts13基因产生的转录物,从而鉴定内含子保留事件。为此目的,我们从thle-3(人肝上皮)细胞的细胞核和细胞质部分中分离出聚a+rna,并使用illumina的truseqstrandedmrna文库制备试剂盒来构建cdna文库。对该文库进行对-端测序,产生了定位到人类基因组的100个核苷酸读数(2009年2月,grch37/hg19组装体)。针对adamts13的测序结果显示在图11中。简而言之,图11示出了使用由ucsc基因组信息学组(centerforbiomolecularscience&engineering,universityofcalifornia,santacruz,1156highstreet,santacruz,ca95064)运行并由例如rosenbloom等人,2015,“theucscgenomebrowserdatabase:2015update,”nucleicacidsresearch43,databaseissue(doi:10.1093/nar/gku1177)描述的ucsc基因组浏览器显现的定位读数,并且可通过峰值信号推测读数的覆盖范围和数目。峰高指示由特定区域中读数的密度给出的表达水平。所有adamts13同种型的示意图(按比例绘制)由ucsc基因组浏览器(读数信号下面)来提供,使得峰值可与adamts13外显子和内含子区域相匹配。根据此显示,我们鉴定了在thle-3细胞的细胞核部分中具有高读数密度,但在这些细胞的细胞质部分中具有极低读数至无读数的两个内含子(25和27,由箭头指示)(如图11的底部图示中的内含子25所示)。这表明这两个内含子均被保留并且含内含子25和内含子27的转录物仍然在细胞核中。这提示这些含保留内含子(ric)的adamts13前mrna是非生产性的,因为它们不向外输出至细胞质。
实施例6:通过adamts13的rnaseq分析所鉴定的内含子保留事件的验证
利用本文所述的方法进行adamts13(血栓性血小板减少性紫癜)基因中内含子25-保留事件的验证(图12)。简而言之,如实施例1中所述采用来自a172(人成胶质细胞瘤)和hepg2(人肝细胞癌)细胞的细胞核和细胞质rna提取物进行放射性逆转录酶pcr(rt-pcr)。在本实施例中,使用导致含内含子25的转录物和正确剪接的转录物二者扩增的位于外显子25和外显子27中的引物来评估内含子保留。产物在5%聚丙烯酰胺凝胶中运行,并通过磷成像进行可视化。以对应于含内含子25的产物的条带强度相对于总转录物(含内含子的加上正确剪接的)的内含子保留百分比(pir)来计算内含子25保留水平。条带的量化表明约80%的adamts13转录物含有内含子25并且该产物保留在细胞核中。此外,放射性rt-pcr结果验证了生物信息学预测,证实rnaseq结果的生物信息学分析是鉴定内含子保留事件的有力工具。
实施例7:靶向adamts13的内含子25的aso步移的设计
将aso步移设计为利用本文所述方法靶向内含子25(图13)。用2’-o-merna(ps主链)、以5个核苷酸间隔移位的18聚体aso(除了1种aso——adam-ivs25-47以外,以避免一段四个鸟嘌呤)靶向内含子255’剪接位点下游紧邻的跨越+6至+58核苷酸的区域和内含子253’剪接位点上游紧邻的跨越-16至-79核苷酸的区域(图13;表4,seqidno:150至167)。根据内含子调节元件集中于剪接位点相邻的序列中的知识选择了这些靶区域。
实施例8:经由adamts13内含子25的aso靶向而提高的剪接效率增加了转录物水平
为了确定我们是否可以通过利用aso提高adamts13内含子25的剪接效率来获得adamts13表达的增加,我们采用了本文所述的方法(图14)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对hepg2细胞进行模拟转染,或用图13和表4中描述的靶向aso,seqidno:150至167中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图14所示)进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+21和+26靶向aso增加了adamts13转录物水平(图14)。将对应于来自靶向-aso转染细胞的adamts13pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自经对照aso处理的细胞的归一化adamts13pcr产物进行绘图。该分析的结果表明,两种靶向aso(+21和+26)使adamts13转录物水平增加将近2.5倍(图14)。这些结果表明,利用aso提高adamts13基因中限速内含子的剪接效率导致基因表达增加。
实施例9:靶向adamts13内含子25的aso的剂量响应效应
为了确定+21和+26aso以及表现出相反效应的-46aso的剂量响应效应(图14),我们采用了本文所述的方法(图15)。利用rnaimax(rim)(invitrogen)递送试剂,如图15所示以浓度渐增的方式对hepg2细胞进行模拟转染,或用三种aso中的每一种或非靶向smn-aso对照独立地转染48小时。放射性rt-pcr结果显示,+21和+26靶向aso与模拟转染或非靶向aso相比增加了adamts13转录物水平,而-46aso与模拟转染或非靶向aso相比则降低了adamts13转录物水平(图15)。将对应于来自靶向-aso转染细胞的adamts13pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自经对照aso处理的细胞的归一化adamts13pcr产物进行绘图。该分析的结果表明,两种靶向aso(+21和+26)使adamts13转录物水平增加将近2.5倍(图15)。这些结果确认,利用aso提高adamts13基因中限速内含子的剪接效率导致基因表达增加。
实施例10:经由adamts13内含子25的aso靶向而提高的剪接效率增加了蛋白质水平
为了检测在用+21或+26aso靶向adamts13内含子25后蛋白质产生量的增加,我们采用了本文所述的方法(图16)。利用rnaimax(rim)(invitrogen)递送试剂,如图16所示以浓度渐增的方式对hepg2细胞进行模拟转染,或用三种aso中的每一种或非靶向smn-aso对照单独转染48小时。简而言之,来自经hepg2处理的细胞的蛋白质提取物在8%sds-聚丙烯酰胺凝胶上运行,并转移到硝酸纤维膜。为了证明蛋白质产生量的增加,采用抗adamts13抗体或抗α微管蛋白抗体分别检测adamts13和作为加样对照的α微管蛋白。图16示出了蛋白质印迹结果,其表明在用+21或+26aso处理后adamts13(顶部图幅)以剂量依赖性的方式增加。将对应于来自靶向-aso转染细胞的adamts13蛋白质的条带强度相对于内源性α微管蛋白进行归一化,并相对于来自模拟处理的细胞的归一化adamts13蛋白质条带进行绘图。该分析的结果表明,靶向aso(+21和+26)使adamts13蛋白水平增加至超过3倍(图16)。这些结果证实,通过采用靶向adamts13内含子25(限速内含子)的5’剪接位点下游的区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所示。
实施例11:靶向adamts13内含子25的+21至+26区域的aso微步移的设计
将aso微步移设计为利用本文所述方法靶向内含子25的+21至+26区域(图17)。用2’-o-me、5’-me-胞嘧啶rna(ps主链)、以1个核苷酸间隔移位的18聚体aso靶向内含子25的5’剪接位点下游的跨越+17至+46的区域(图17;表4,seqidno:184至197)。根据所观察到的aso+21和+26的效果(图16)来选择靶区域。
实施例12:经由adamts13内含子25+21至+26区域的aso微步移靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用微步移aso提高adamts13内含子25的剪接效率来获得adamts13表达的增加,我们采用了本文所述的方法(图18)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对hepg2细胞进行模拟转染,或用图17以及表4中seqidno:184至197所描述的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图18所示)进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso以及两种原始的+21和+26aso(淡灰色柱条,图18)相比,具有5’-me-胞嘧啶的+21和+25靶向aso进一步增加了adamts13转录物水平。将对应于来自靶向-aso转染细胞的adamts13pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自经对照aso处理的细胞的归一化adamts13pcr产物进行绘图。该分析的结果表明,两种靶向aso(+21和+25)使adamts13转录物水平增加至将近2.0倍(图18)。这些结果表明,利用aso提高adamts13基因中限速内含子的剪接效率导致了基因表达的增加,并且通过微步移精修靶区域可导致鉴定更有效的aso。
实施例13:使用新一代测序通过rnaseq鉴定tsc1转录物中的内含子保留事件
我们使用新一代测序进行全转录物组鸟枪法测序,以概略显示由tsc1基因产生的转录物,从而鉴定内含子保留事件。为此,我们从原代人星形细胞(ast)和原代人皮质神经元(hcn)细胞的细胞核和细胞质部分中分离出聚a+rna,并使用illumina的truseqstrandedmrna文库制备试剂盒构建cdna文库。对该文库进行对-端测序,产生了定位到人类基因组的100个核苷酸的读数(2009年2月,grch37/hg19组装体)。tsc1的测序结果显示在图19中。简而言之,图19显示了利用ucsc基因组浏览器所显现的定位读数,并且读数的覆盖范围和数目可通过峰值信号来推测。峰高指示由特定区域中读数的密度给出的表达水平。所有tsc1同种型的示意图(按比例绘制)由ucsc基因组浏览器(读数信号下面)提供,使得峰值可与tsc1外显子和内含子区域相匹配。根据此显示,我们鉴定了在ast和hcn细胞的细胞核部分中具有高读数密度,但在这些细胞的细胞质部分中具有极低读数至无读数的三个内含子(5、10和11,由箭头指示)(如图19的底部图示中的内含子10所示)。这表明两个内含子均被保留,并且含内含子5、内含子10和内含子11的转录物仍然在细胞核中。这提示这些含保留内含子(ric)的tsc1前mrna是非生产性的,因为它们未向外输出至细胞质。
实施例14:通过tsc1的rnaseq分析所鉴定的内含子保留事件的验证
利用本文所述的方法进行tsc1(复合型结节性硬化症1)基因中内含子10-保留事件的验证(图20)。简而言之,如实施例1中所述利用来自a172(人成胶质细胞瘤)细胞的细胞核和细胞质rna提取物进行放射性逆转录酶pcr(rt-pcr)。在该实施例中,利用导致含内含子10的转录物和正确剪接的转录物均扩增的位于外显子9和外显子11中的引物来评估内含子保留。产物在5%聚丙烯酰胺凝胶中运行,并通过磷成像进行可视化。以对应于含内含子10的产物的条带强度相对于总转录物(含内含子的加上正确剪接的)的内含子保留百分比(pir)来计算内含子10保留水平。条带的量化表明约36%的tsc1转录物含有内含子10并且该产物保留在细胞核中。此外,放射性rt-pcr结果验证了生物信息学预测,证实rnaseq结果的生物信息学分析是鉴定内含子保留事件的有力工具。
实施例15:靶向tsc1的内含子10的aso步移的设计
将aso步移设计为利用本文所述方法靶向内含子10(图21)。用2’-o-merna(ps主链)、以5个核苷酸间隔移位的18聚体aso靶向内含子10的5’剪接位点下游紧邻的跨越+6至+58核苷酸的区域和内含子10的3’剪接位点上游紧邻的跨越-16至-68核苷酸的区域(图21;表5,seqidno:214至229)。根据内含子调节元件集中于剪接位点相邻的序列中的知识选择了这些靶区域。
实施例16:经由tsc1内含子10的aso靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用aso提高tsc1内含子10的剪接效率来获得tsc1表达的增加,我们采用了本文所述的方法(图22)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对a172细胞进行模拟转染,或用图21以及表5中seqidno:214至229所描述的的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图22所示)进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+31靶向aso增加了tsc1转录物水平(图22)。将对应于来自靶向-aso转染细胞的tsc1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化tsc1pcr产物进行绘图。该分析的结果表明,若干种aso(包括+31)使tsc1转录物水平增加至将近1.5倍(图22)。这些结果表明,利用aso提高tsc1基因中限速内含子的剪接效率导致基因表达增加。
实施例17:靶向tsc1内含子10的aso的剂量响应效应
为了确定+31aso的剂量响应效应,我们采用了本文所述的方法(图23)。使用rnaimax(rim)(invitrogen)递送试剂,如图23所示以浓度渐增的方式对a172细胞进行模拟转染或用+31aso或非靶向smn-aso对照独立转染达72小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+31靶向aso增加了tsc1转录物水平(图23)。将对应于来自靶向-aso转染细胞的tsc1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化tsc1pcr产物进行绘图。该分析的结果表明,+31靶向aso使tsc1转录物水平以剂量依赖性的方式增加至将近2.0倍(图23)。利用tsc1转录物中别处的引物通过rtqpcr确认了这些结果,显示了至3倍的增加以及对aso处理的剂量依赖性响应。这些结果确认,利用aso提高tsc1基因中限速内含子的剪接效率导致基因表达增加。
实施例18:经由tsc1内含子10的aso靶向而提高的剪接效率增加蛋白质水平
为了检测在用+31aso靶向tsc1内含子10后蛋白质产生量的增加,我们采用了本文所述的方法(图24)。利用rnaimax(rim)(invitrogen)递送试剂,如图24所示以浓度渐增的方式对a172细胞进行模拟转染或用+31aso或非靶向smn-aso对照独立转染72小时。简而言之,来自经a172处理的细胞的蛋白质提取物在10%sds-聚丙烯酰胺凝胶上运行,并转移到硝酸纤维膜。为了证明蛋白质产生量的增加,采用抗tsc1抗体或抗α微管蛋白抗体分别检测tsc1和作为加样对照的α微管蛋白。图24显示了蛋白质印迹结果,其表明在用30和60nm的+31aso处理后tsc1(顶部图幅)以剂量依赖性的方式增加。将对应于来自靶向-aso转染细胞的tsc1蛋白的条带强度相对于内源性α微管蛋白进行归一化,并相对于来自模拟处理的细胞的归一化tsc1蛋白条带进行绘图。该分析的结果表明,靶向aso(+31)使tsc1蛋白水平增加超过2倍(图24)。这些结果证实,通过利用靶向tsc1内含子10(限速内含子)的5’剪接位点下游的区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所示。
实施例19:利用新一代测序通过rnaseq鉴定impdh1转录物中的内含子保留事件
我们利用新一代测序进行全转录物组鸟枪法测序,以概略显示由impdh1基因(10型视网膜色素变性)产生的转录物,从而鉴定内含子保留事件。为此目的,我们从arpe-19(人视网膜上皮)细胞的细胞核和细胞质部分中分离出聚a+rna,并利用illumina的truseqstrandedmrna文库制备试剂盒来构建cdna文库。对该文库进行对-端测序,产生了定位到人类基因组的100个核苷酸的读数(2009年2月,grch37/hg19组装体)。impdh1的测序结果显示在图25中。简而言之,图25显示了利用ucsc基因组浏览器显现的定位读数,并且读数的覆盖范围和数目可通过峰值信号来推断。峰高指示由特定区域中读数的密度给出的表达水平。所有impdh1同种型的示意图(按比例绘制)由ucsc基因组浏览器(读数信号下面)来提供,使得峰值可与impdh1外显子和内含子区域相匹配。根据此显示,我们鉴定了在arpe-19细胞的细胞核部分中具有高读数密度,但在这些细胞的细胞质部分中没有读数的一个内含子(14,由箭头指示)(如图25的底部图示中的内含子14所示)。这表明内含子14被保留并且含内含子14的转录物仍然在细胞核中。这提示含保留内含子(ric)的impdh1前mrna是非生产性的,因为它未向外输出至细胞质。
实施例20:靶向impdh1的内含子14的aso步移的设计
将aso步移设计为利用本文所述方法靶向内含子14(图26)。用2’-o-merna(ps主链)、以5个核苷酸间隔移位的18聚体aso(除了1种aso——imp-ivs14+18以外,以避免一段四个鸟嘌呤)来靶向内含子14的5’剪接位点下游紧邻的跨越+6至+65核苷酸的区域和内含子14的3’剪接位点上游紧邻的跨越-16至-68核苷酸的区域(图26;表6,seqidno:246至261)。根据内含子调节元件集中于剪接位点相邻的序列中的知识选择了这些靶区域。
实施例21:经由impdh1内含子14的aso靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用aso提高impdh1内含子14的剪接效率来获得impdh1表达的增加,我们采用了本文所述的方法(图27)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对arpe-19细胞进行模拟转染,或用图26和表6中seqidno:246至261所描述的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图27所示)进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+48靶向aso增加了impdh1转录物水平(图27)。将对应于来自靶向-aso转染细胞的impdh1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自经对照aso处理的细胞的归一化impdh1pcr产物进行绘图。该分析的结果表明,靶向aso(+48)使impdh1转录物水平增加4.0倍(图27)。这些结果表明,利用aso提高impdh1基因中限速内含子的剪接效率导致基因表达增加。
实施例22:靶向impdh1内含子14的aso+48的剂量响应效应
为了确定+48aso的剂量响应效应,我们采用了本文所述的方法(图28)。利用rnaimax(rim)(invitrogen)递送试剂,如图28所示以浓度渐增的方式对arpe-19细胞进行模拟转染或用+48aso或非靶向smn-aso对照独立转染72小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+48靶向aso以剂量依赖性的方式增加了impdh1转录物水平(图28)。将对应于来自靶向-aso转染细胞的impdh1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化impdh1pcr产物进行绘图。该分析的结果表明,靶向aso(+48)使impdh1转录物水平增加将近1.5倍(图28,中间图)。利用impdh1转录物中别处的引物通过rtqpcr证实了这些结果,显示了2.5倍的增加以及对aso处理的剂量依赖性响应(图28,右图)。此外,对于内含子14保留计算pir(如实施例6所述)并对所述值进行绘图,其表明随着aso浓度和正确剪接的转录物增加,观察到内含子14保留的减少(图28,左图)。这些结果确认,利用aso提高impdh1基因中限速内含子的剪接效率导致基因表达增加。
实施例23:经由impdh1内含子14的aso靶向而提高的剪接效率增加蛋白质水平
为了检测在用+48aso靶向impdh1内含子14后蛋白质产生量的增加,我们采用了本文所述的方法(图29)。利用rnaimax(rim)(invitrogen)递送试剂,如图29所示以浓度渐增的方式对arpe-19细胞进行模拟转染或用+48aso或非靶向smn-aso对照独立转染72小时。简而言之,来自经arpe-19处理的细胞的蛋白质提取物在4-20%sds-聚丙烯酰胺凝胶上运行,并转移到硝酸纤维膜。为了证明蛋白质产生量的增加,采用抗impdh1抗体、抗β连环蛋白抗体或β肌动蛋白分别检测impdh1,和作为加样对照的β连环蛋白或β肌动蛋白。图29显示了蛋白质印迹结果,其表明在用+48aso处理后impdh1以剂量依赖性的方式增加。将对应于来自靶向-aso转染细胞的impdh1蛋白的条带强度相对于内源性β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化impdh1蛋白条带进行绘图。该分析的结果表明,靶向aso(+48)使impdh1蛋白水平增加至将近2.5倍(图29)。这些结果证实,通过利用靶向impdh1内含子14(限速内含子)的5’剪接位点下游的区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所示。
实施例24:靶向impdh1内含子14的+48区域的aso微步移的设计
将aso微步移设计为利用本文所述方法靶向内含子14的+44至+70区域(图30)。用2’-o-me、5’-me-胞嘧啶rna(ps主链)、以1个核苷酸间隔移位的18聚体aso靶向内含子14的5’剪接位点下游的跨越+44至+70的区域(图30;表6,seqidno:262至271)。根据所观察到的aso+48的效果(图29)选择了靶区域。
实施例25:经由impdh1内含子14+48区域的aso微步移靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用微步移aso提高impdh1内含子14的剪接效率来获得impdh1表达的增加,我们采用了本文所述的方法(图31)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对arpe-19细胞进行模拟转染,或用图30和表6seqidno:262至271中所描述的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图31所示)进行实验达48小时。rt-qpcr结果显示,与模拟转染或非靶向aso以及原始的+48aso相比,+46和+47靶向aso进一步增加了impdh1转录物水平(图31)。该分析的结果表明,两种靶向aso(+46和+47)使impdh1转录物水平增加至超过3.0倍(图31)。这些结果表明,利用aso提高impdh1基因中限速内含子的剪接速度导致了基因表达的增加,并且通过微步移精修靶区域可导致鉴定更有效的aso。
实施例26:利用新一代测序通过rnaseq鉴定pkd1转录物中的内含子保留事件
我们利用新一代测序进行全转录物组鸟枪法测序,以概略显示由pkd1基因(多囊肾病)产生的转录物,从而鉴定内含子保留事件。为此,我们从原代人肾上皮(ren)细胞的细胞核和细胞质部分中分离出聚a+rna,并利用illumina的truseqstrandedmrna文库制备试剂盒来构建cdna文库。对该文库进行对-端测序,产生了定位到人类基因组的100个核苷酸的读数(2009年2月,grch37/hg19组装体)。pkd1的测序结果显示在图32中。简而言之,图32显示了利用ucsc基因组浏览器显现的定位读数,并且读数的覆盖范围和数目可通过峰值信号加以推断。峰高指示由特定区域中读数的密度给出的表达水平。所有pkd1同种型的示意图(按比例绘制)由ucsc基因组浏览器(读数信号下面)提供,使得峰值可与pkd1外显子和内含子区域相匹配。根据此显示,我们鉴定了在ren细胞的细胞核部分中具有高读数密度,但在这些细胞的细胞质部分中具有极低读数至无读数的四个内含子(32、33、37和38,由箭头指示)(如图32的底部图示中的内含子37所示)。这表明这四个内含子均被保留并且含内含子32、内含子33、内含子37和内含子38的转录物仍然在细胞核中。这提示这些含保留内含子(ric)的pkd1前mrna是非生产性的,因为它们未向外输出至细胞质。
实施例27:靶向pkd1的内含子37的aso步移的设计
将aso步移设计为利用本文所述方法靶向内含子37(图33)。用2’-o-merna(ps主链)、以5个核苷酸间隔移位的18聚体aso(除了2种aso——pkd1-ivs37+8和+24以外,以避免一段四个鸟嘌呤)靶向内含子375’剪接位点下游紧邻的跨越+6至+66核苷酸的区域和内含子373’剪接位点上游紧邻的跨越-16至-51核苷酸的区域(图33;表7,seqidno:297至312)。根据内含子调节元件集中于剪接位点相邻的序列中的知识选择了这些靶区域。
实施例28:经由pkd1内含子37的aso靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用aso提高pkd1内含子37的剪接效率来获得pkd1表达的增加,我们采用了本文所述的方法(图34)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对hek293细胞进行模拟转染,或用图33和表7中seqidno:297至312所描述的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图34所示)进行实验达48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+29靶向aso增加了pkd1转录物水平(图34)。将对应于来自靶向-aso转染细胞的pkd1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化pkd1pcr产物进行绘图。来自该分析的结果表明,+29aso使pkd1转录物水平增加至1.8倍(图34)。这些结果表明,利用aso提高pkd1基因中限速内含子的剪接效率导致基因表达增加。
实施例29:靶向pkd1内含子37的aso的剂量响应效应
为了确定+29aso的剂量-响应效应,我们采用了本文所述的方法(图35)。利用rnaimax(rim)(invitrogen)递送试剂,如图35所示以浓度渐增的方式对hek293细胞进行模拟转染或用+29aso或非靶向smn-aso对照独立转染48小时。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,+29靶向aso增加了pkd1转录物水平(图35)。将对应于来自靶向-aso转染细胞的pkd1pcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化pkd1pcr产物进行绘图。该分析的结果表明,+29靶向aso使pkd1转录物水平以剂量依赖性的方式增加至超过2.0倍(图35,中间图)。使用pkd1转录物中别处的引物通过rtqpcr确认了这些结果,显示了超过2倍的增加以及对aso处理的剂量依赖性响应(图35,右图)。此外,对于内含子37保留计算pir(如实施例6所述)并对所述值进行绘图,其表明随着aso浓度和正确剪接的转录物增加,观察到内含子37保留的减少(图35,左图)。这些结果确认,利用aso提高pkd1基因中限速内含子的剪接效率导致基因表达增加。
实施例30:经由pkd1内含子37的aso靶向而提高的剪接效率增加蛋白质水平
为了检测在用+29aso靶向pkd1内含子37后蛋白质产生量的增加,我们采用了本文所述的方法(图36)。利用rnaimax(rim)(invitrogen)递送试剂,如图36所示以浓度渐增的方式对hek293细胞进行模拟转染或用+29aso或非靶向smn-aso对照独立转染72小时。简而言之,将细胞固定和透化处理,并用抗pkd1抗体或igg同种型对照抗体进行处理。通过流式细胞术计数10,000个细胞来分析细胞。图36显示了荧光强度/细胞计数的图,其表明与模拟处理(未处理)的细胞相比,aso浓度越高的细胞具有越强的pkd1信号。由对应于经+29aso处理的细胞的荧光强度相对于对应于模拟处理的细胞的荧光强度的倍数变化进行绘图。该分析的结果表明,靶向aso(+29)使pkd1蛋白水平增加至将近1.5倍(图36)。这些结果证实,通过利用靶向pkd1内含子37(限速内含子)的5’剪接位点下游的区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所示。
实施例31:利用新一代测序通过rnaseq鉴定ikbkap转录物中的内含子保留事件
我们利用新一代测序进行全转录物组鸟枪法测序,以概略显示由ikbkap基因产生的转录物,从而鉴定内含子保留事件。为此目的,我们从arpe-19、ast、人支气管上皮(bron)、hcn、ren和thle-3细胞的细胞核和细胞质部分中分离出聚a+rna,并利用illumina的truseqstrandedmrna文库制备试剂盒来构建cdna文库。对该文库进行对-端测序,产生了定位到人类基因组的100个核苷酸的读数(2009年2月,grch37/hg19组装体)。ikbkap的测序结果显示在图37中。简而言之,图37显示了利用ucsc基因组浏览器显现的定位读数,并且读数的覆盖范围和数目可通过峰值信号加以推断。峰高指示由特定区域中读数的密度给出的表达水平。所有ikbkap同种型的示意图(按比例绘制)由ucsc基因组浏览器(读数信号下面)提供,使得峰值可与ikbkap外显子和内含子区域相匹配。根据此显示,我们鉴定了在所有测序细胞的细胞核部分中具有高读数密度,但在这些细胞的细胞质部分中没有读数的2个内含子(7和8,由箭头指示)(如图37的底部图示中的两个内含子所示)。这表明内含子7和8均被保留并且含内含子7和内含子8的转录物仍然在细胞核中。这提示含保留内含子(ric)的ikbkap前mrna是非生产性的,因为它们未向外输出至细胞质。
实施例32:通过ikbkap的rnaseq分析鉴定的内含子保留事件的验证
利用本文所述的方法进行ikbkap(家族性自主神经功能异常)基因中内含子7-保留事件的验证(图38)。简而言之,如实施例1中所述采用来自arpe-19、hela和u2os细胞的细胞核和细胞质rna提取物进行放射性逆转录酶pcr(rt-pcr)。在该实施例中,利用导致含内含子7的转录物和正确剪接的转录物均扩增的位于外显子6和外显子8中的引物来评估内含子保留。产物在5%聚丙烯酰胺凝胶中运行,并通过磷成像进行可视化。以对应于含内含子7的产物的条带强度相对于总转录物(含内含子的加上正确剪接的)的内含子保留百分比(pir)来计算内含子7保留水平。条带的量化表明约35%的ikbkap转录物含有内含子7并且该产物保留在细胞核中。此外,放射性rt-pcr结果验证了生物信息学预测,证实rnaseq结果的生物信息学分析是鉴定内含子保留事件的有力工具。
实施例33:靶向ikbkap的内含子7和8的aso步移的设计
将aso步移设计为利用本文所述方法靶向内含子7(顶部图幅)或内含子8(底部图幅)(图39)。用2’-o-merna(ps主链)、以5个核苷酸间隔移位的18聚体aso靶向内含子7或8的5’剪接位点下游紧邻的跨越+6至+58核苷酸的区域和内含子7或8的3’剪接位点上游紧邻的跨越-16至-68核苷酸的区域(图39;表8,seqidno:329至360)。根据内含子调节元件集中于剪接位点相邻的序列中的知识选择了这些靶区域。
实施例34:经由ikbkap内含子7和8的aso靶向而提高的剪接效率增加转录物水平
为了确定我们是否可以通过利用aso提高ikbkap内含子7或8的剪接效率来获得ikbkap表达的增加,我们采用了本文所述的方法(图40)。为此,利用rnaimax(rim)(invitrogen)递送试剂,对arpe-19细胞进行模拟转染,或用图39和表8中seqidno:329至360所描述的靶向aso中的每一种或非靶向smn-aso对照独立地进行转染。利用60nmaso(如图40所示)进行实验达48小时。相对于来自模拟处理的细胞的归一化ikbkappcr产物而绘图的rt-qpcr结果显示,与模拟转染或非靶向aso相比,ivs7+26靶向aso(顶部图)以及ivs8+26和-16(底部图)靶向aso增加了ikbkap转录物水平(图40)。该分析表明,这些aso使ikbkap转录物水平增加将近1.2-1.6倍(图40)。这些结果表明,利用aso提高ikbkap基因中限速内含子的剪接效率导致基因表达增加。
实施例35:靶向ikbkap内含子7和8的aso的剂量响应效应
为了确定ivs7+26和ivs8-16aso的剂量-响应效应,我们采用了本文所述的方法(图41)。利用rnaimax(rim)(invitrogen)递送试剂,以浓度渐增的方式对arpe-19细胞进行模拟转染,或用ivs7+26或ivs8-16aso,或非靶向smn-aso对照独立地进行转染,或用各自45nm的两种aso的组合转染72小时(图41)。放射性rt-pcr结果显示,与模拟转染或非靶向aso相比,ivs7+26或ivs8-16靶向aso以剂量依赖性的方式增加了ikbkap转录物水平(图41)。将对应于来自靶向-aso转染细胞的ikbkappcr产物的条带强度相对于β肌动蛋白进行归一化,并相对于来自模拟处理的细胞的归一化ikbkappcr产物进行绘图。该分析的结果表明,ivs7+26和ivs8-16靶向aso,并且它们的组合使ikbkap转录物水平以剂量依赖性的方式增加至2.0-2.5倍(图40)。这些结果确认,利用aso提高ikbkap基因中限速内含子的剪接效率导致基因表达增加。
实施例36:经由ikbkap内含子7或8的aso靶向而提高的剪接效率增加蛋白质水平
为了分别检测在用ivs7+26aso或ivs8-16aso靶向ikbkap内含子7或8后蛋白质产生量的增加,我们采用了本文所述的方法(图42)。利用rnaimax(rim)(invitrogen)递送试剂,以浓度渐增的方式对arpe-19细胞进行模拟转染,或用ivs7+26aso或ivs8-16aso,或非靶向smn-aso对照独立地进行转染,或用各自45nm的两种aso的组合转染72小时(图42)。简而言之,来自经arpe-19处理的细胞的蛋白质提取物在4-20%sds-聚丙烯酰胺凝胶上运行,并转移到硝酸纤维膜。为了证明蛋白质产生量的增加,采用抗ikap抗体或抗β连环蛋白抗体分别检测ikap,和作为加样对照的β连环蛋白。图42显示了蛋白质印迹结果,其表明在用ivs7+26aso或ivs8-16aso或两种aso的组合处理后ikap以剂量依赖性的方式增加。将对应于来自靶向-aso转染细胞的ikap蛋白的条带强度相对于内源性β连环蛋白进行归一化,并相对于来自模拟处理的细胞的归一化ikap蛋白质条带进行绘图。该分析的结果表明,靶向asoivs7+26和ivs8-16使ikap蛋白水平增加至约3倍(图42)。这些结果证实,通过利用靶向ikbkap内含子7的5’剪接位点下游的区域或ikbkap内含子8的3’剪接位点上游的区域的aso来提升剪接效率导致了靶蛋白产生量的增加,如图2所述。
表11:prpf31靶序列
表12:rb1靶序列
表13:hbb靶序列
表14:hbg1/hbg2靶序列
表15:cftr靶序列
表16:adamts13靶序列
表17:tsc1靶序列
表18:impdh1靶序列
表19:pkd1靶序列
表20:ikbkap靶序列
- 还没有人留言评论。精彩留言会获得点赞!