5‑取代‑4‑溴‑1H‑吡咯‑2‑甲酸酯类化合物及其制备方法与流程
本发明属于药物制备
技术领域:
,具体涉及一种吡唑衍生物及其制备方法。
背景技术:
:吡咯衍生物广泛地存在于自然界中,在生命体的新陈代谢中起着及其重要的作用。目前,含吡咯环类化合物已广泛用于抑菌、抑酶及治疗心血管疾病等方面的研究与应用。而ɑ-吡咯甲酰基衍生物已用于合成多种生理活性碱,如具有显著抗肿瘤活性及NCI60菌株有良好抑制作用的Dibro,以及对蛋白激酶有抑制作用的Axinohydantoins。技术实现要素:本发明目的在于提供一种如式(I)的5-取代-4-溴-1H-吡咯-2-甲酸酯及其制备方法,及用于制备低毒高效,抑菌抑酶,消炎镇痛,及治疗心血管疾病的化合物及其合成中间体。一种吡唑衍生物,化学名称5-取代-4-溴-1H-吡咯-2-甲酸酯,如式(I)所示,其中,R1选自-CH3、-C2H5,R2选自-Cl、-I、-CF2、-CF3、-CN、-CH2OH、-CHO,-COOH、-CH2NHC6H4Cl、-CH2OCOCH2CH3、-CH2OCOC6H4CH3、-COOCH2CH3、-COOC6H5、-CONHC6H4Cl。本发明的目的在于提供一种所述式(I)的化合物的制备方法,包括以下步骤:(A)2-三氯乙酰基吡咯的制备:向反应瓶中加入无水乙醚和式(I)的吡咯,并向其中缓慢滴加无水乙醚和三氯乙酰氯的混合液,继续搅拌回流,然后用饱和碳酸钠溶液淬灭反应,收集有机相,干燥脱色后,减压蒸除溶剂后得到白色粉末状固体化合物(2)的2-三氯乙酰基吡咯,反应式如下:(B)向反应烧瓶中加入2-三氯乙酰基吡咯和乙腈,控制温度为0℃~-25℃,然后向三颈烧瓶中慢慢加入NBS,停止反应后出现白色不溶物,抽滤,滤饼用水淋洗,向滤液中加入冰水后,出现大量紫灰色固体,抽滤,将得到的滤饼干燥,并用乙醇和水混合溶剂重结晶得到白色固体化合物(3)的4-溴-2-三氯乙酰基吡咯,反应式如下:(C)向反应烧瓶中加入金属钠和乙醇,待金属钠完全溶解后,烧瓶内温度降至10℃,取化合物4-溴-2-三氯乙酰基吡咯和无水乙醇,溶解后缓慢滴加到三颈烧瓶内,室温搅拌,反应结束后减压蒸除溶剂,然后向所得的残渣中加入冰乙醇和水,体积比为2:1,混合搅拌,抽滤得橘红色固体化合物(4)的4-溴-1H-吡咯-2-甲酸乙酯,反应式如下:(D)以化合物(4)4-溴-1H-吡咯-2-甲酸乙酯为原料,直接对5位取代,制备得到式(I)化合物,或者通过5位醛基进行一系列转化,制备得到式(I)5-取代-4-溴-1H-吡咯-2-甲酸酯,其中,R1选自-CH3、-C2H5,R2选自-Cl、-I、-CF2、-CF3、-CN、-CH2OH、-CHO,-COOH、-CH2NHC6H4Cl、-CH2OCOCH2CH3、-CH2OCOC6H4CH3、-COOCH2CH3、-COOC6H5、-CONHC6H4Cl。优选的化合物(4)与NCS在醋酸溶液中,反应生成化合物(5)优选的化合物(4)与I2、C2AgF3O2在二氯甲烷溶液中,反应生成化合物(6)优选的化合物(4)与CF3SO2Cl在二氯甲烷溶液中,反应生成化合物(7)优选的化合物(4)与氯磺酰异氰酸酯在乙腈溶液中,反应生成化合物(8)优选的化合物(4)与DMF、POCl3在二氯甲烷溶液中,反应生成化合物(9)优选的所述化合物(9)与四氯苯胺缩合,再与NaBH4还原反应生成化合物(11)所述化合物(9)与NaBH4还原反应生成化合物(12)所述化合物(9)与亚氯酸钠氧化反应生成化合物(15)优选的所述化合物(12)与乙酰氯反应生成化合物(13)优选的所述化合物(12)与对甲基苯甲酰氯反应生成化合物(14)优选的所述化合物(15)与氯化亚砜的乙醇溶液反应生成化合物(16)优选的化合物(15)与草酰氯反应,再与苯酚反应生成化合物(17)优选的化合物(15)与草酰氯反应,再与4-氯苯胺反应生成化合物(18)本发明的另一目的在于提供一种药物组合物,所述药物组合物包含所述的式(I)化合物以及药学上可接受的载体或稀释剂。本发明的另一目的在于提供一种所述的式(I)化合物用于制备治疗心血管疾病的药物的用途。本发明的另一目的在于提供一种所述的式(I)化合物用于制备抑菌抑酶药物的用途。本发明的另一目的在于提供一种所述的式(I)化合物用于制备消炎镇痛药物的用途。本发明提供了一种制备通式(Ⅰ)的化合物的方法:其中,R1选自-CH3、-C2H5,R2选自-Cl、-I、-CF2、-CF3、-CN、-CH2OH、-CHO,-COOH、-CH2NHC6H4Cl、-CH2OCOCH2CH3、-CH2OCOC6H4CH3、-COOCH2CH3、-COOC6H5、-CONHC6H4Cl;其包括如下步骤:A以化合物(4)为基础原料合成各种5-取代-4-溴-1H-吡咯-2-甲酸酯类化合物i.化合物(4)与NCS在醋酸溶液中,反应生成化合物(5)ii.化合物(4)与I2、C2AgF3O2在二氯甲烷溶液中,反应生成化合物(6)iii.化合物(4)与CF3SO2Cl在二氯甲烷溶液中,反应生成化合物(7)iv.化合物(4)与氯磺酰异氰酸酯在乙腈溶液中,生成化合物(8)B以化合物(9)为基础原料合成各种5-取代-4-溴-1H-吡咯-2-甲酸酯类化合物i.化合物(9)与DAST在二氯甲烷溶液中,反应生成化合物(10)ii.化合物(9)与四氯苯胺缩合,在NaBH4还原反应生成化合物(11)iii.化合物(9)在NaBH4还原反应生成化合物(12)iv.化合物(12)与乙酰氯反应,生成化合物(13)v.化合物(12)与对甲基苯甲酰氯反应,生成化合物(14)vi.化合物(9)在亚氯酸钠氧化反应生成化合物(15)vii.化合物(15)在氯化亚砜的乙醇溶液,反应生成化合物(16)viii.化合物(15)先与草酰氯反应,在与苯酚反应生成化合物(17)ix.化合物(15)与草酰氯反应,再与四氯苯胺反应生成化合物(18)以上各种合成的化合物即为通式(Ⅰ)化合物,其中:产生通式(Ⅰ)化合物的方法可分成两种,一种是以化合物(4)为原料的合成方法A,一种是以化合物(10)为原料的合成方法B;其中方法A是直接对5位取代,制备化合物;方法B则是通过5位醛基进行一系列转化,以制备化合物,每个取代基R2是独立的官能团或者取代物。附图说明下面结合附图及实施例对本发明作进一步描述:图1为化合物(5)的核磁共振氢谱,图2为化合物(5)的核磁共振碳谱,图3为化合物(6)的核磁共振氢谱,图4为化合物(6)的核磁共振碳谱,图5为化合物(7)的核磁共振氢谱,图6为化合物(7)的核磁共振碳谱,图7为化合物(8)的核磁共振氢谱,图8为化合物(8)的核磁共振碳谱,图9为化合物(9)的核磁共振氢谱,图10为化合物(9)的核磁共振碳谱,图11为化合物(10)的核磁共振氢谱,图12为化合物(10)的核磁共振碳谱,图13为化合物(11)的核磁共振氢谱,图14为化合物(11)的核磁共振碳谱,图15为化合物(12)的核磁共振氢谱,图16为化合物(12)的核磁共振碳谱,图17为化合物(13)的核磁共振氢谱,图18为化合物(13)的核磁共振碳谱,图19为化合物(14)的核磁共振氢谱,图20为化合物(14)的核磁共振碳谱,图21为化合物(15)的核磁共振氢谱,图22为化合物(15)的核磁共振碳谱,图23为化合物(16)的核磁共振氢谱,图24为化合物(16)的核磁共振碳谱,图25为化合物(17)的核磁共振氢谱,图26为化合物(17)的核磁共振碳谱,图27为化合物(18)的核磁共振氢谱,图28为化合物(18)的核磁共振碳谱。具体实施方式定义如本发明的说明书中所用的,以下的词语和短语通常被认为具有如下阐明的含说明书义,除非在使用这些词语或短语的上下文中另有指明。如本文中使用的术语“烷基”是指具有所示数目的碳原子的直链或支链饱和烃。例如,(C1-C8)烷基表示包括,但不限于甲基、乙基、丙基、异丙基、丁基、仲-丁基、叔-丁基、戊基、异戊基、新戊基、己基、异己基和新己基。烷基基团可以是未取代的或可选被一个或多个本文中所述的取代基取代。术语“三氯乙酰基”是指基团三氯乙酰基-COOCCl3,术语“4-溴-2-三氯乙酰基吡咯”中的溴是指取代基团溴取代基-Br,术语“4-溴-1H-吡咯-2-甲酸乙酯”中的甲酸乙酯是指基团甲酸乙酯基团-COOC2H5,术语“5-醛基-4-溴-1H-吡咯-2-甲酸乙酯”中的醛基是指基团醛基-CHO,术语“5-氟-4-溴-1H-吡咯-2-甲酸乙酯”中的氟是指取代基氟-F,术语“5-碘-4-溴-1H-吡咯-2-甲酸乙酯”中的碘是指取代基碘-I,术语“5-氰基-4-溴-1H-吡咯-2-甲酸乙酯”中的氰基是指取代基氰基-CN,术语“5-二氟甲基-4-溴-1H-吡咯-2-甲酸乙酯”中的二氟甲基是指取代基团二氟甲基-CF2,术语“5-三氟甲基-4-溴-1H-吡咯-2-甲酸乙酯”中的三氟甲基是指取代基团三氟甲基-CF3,术语“5-苄醇-4-溴-1H-吡咯-2-甲酸乙酯”中的苄醇是指取代基团亚甲羟基-CH2OH,术语“5-羧基-4-溴-1H-吡咯-2-甲酸乙酯”中的羧基是指取代基团羧基-COOH,术语“5-(4-氯-N甲基苯胺)-4-溴-1H-吡咯-2-甲酸乙酯”中的4-氯-N-甲基苯胺是指取代基4-氯-N-甲基苯胺-CH2NHC6H4Cl,术语“5-(乙酸甲酯)-4-溴-1H-吡咯-2-甲酸乙酯”中的乙酸甲酯是指取代基团乙酸己酯-CH2OOCCH3,术语“5-(对甲基苯甲酸甲酯)-4-溴-1H-吡咯-2-甲酸乙酯”中的对甲基苯甲酸甲酯是指取代基团对甲基苯甲酸甲酯-CH2OOCC6H4CH3,术语“5-(甲酸乙酯)-4-溴-1H-吡咯-2-甲酸乙酯”中的甲酸乙酯是指取代基团甲酸乙酯-COOC2H5,术语“5-(甲酸苯酯)-4-溴-1H-吡咯-2-甲酸乙酯”中的甲酸苯酯是指取代基团甲酸苯酯-COOC6H5,术语“5-[N-(4-氯苯基)甲酰胺]-4-溴-1H-吡咯-2-甲酸乙酯”中的N-(4-氯苯基)甲酰胺是指取代基N-(4-氯苯基)甲酰胺-CONHC6H4Cl,术语“羟基(hydroxy)”或“羟基(hydroxyl)”是指基团-OH。此外,本文中使用的缩写具有如下各自的含义:NBSN-溴代丁二酰亚胺NCSN-氯代丁二酰亚胺DMFN,N-二甲基甲酰胺I2单质碘POCl3三氯氧磷selectflour1-氯甲基-4-氟-1,4-重氮化二环2.2.2辛烷双(四氟硼酸)盐DAST二乙胺基三氟化硫H2O2双氧水DMSO二甲基亚砜DMAP4-二甲氨基吡啶FeSO4·7H2O硫酸亚铁其中所述CF3SO2Cl是三氟甲磺酰氯,所述Et是指乙基即-C2H5基团。药物组合物术语“药物组合物”包括适于给予哺乳动物(例如人)的制剂。当本发明化合物是作为药物给予哺乳动物(例如人)时,它们可以自身的形式或者以含有例如0.1%至99.9%(更优选地0.5至90%)活性成分连同药学上可接受载体的药物组合物的形式而给予。术语“药学上可接受的载体”是本
技术领域:
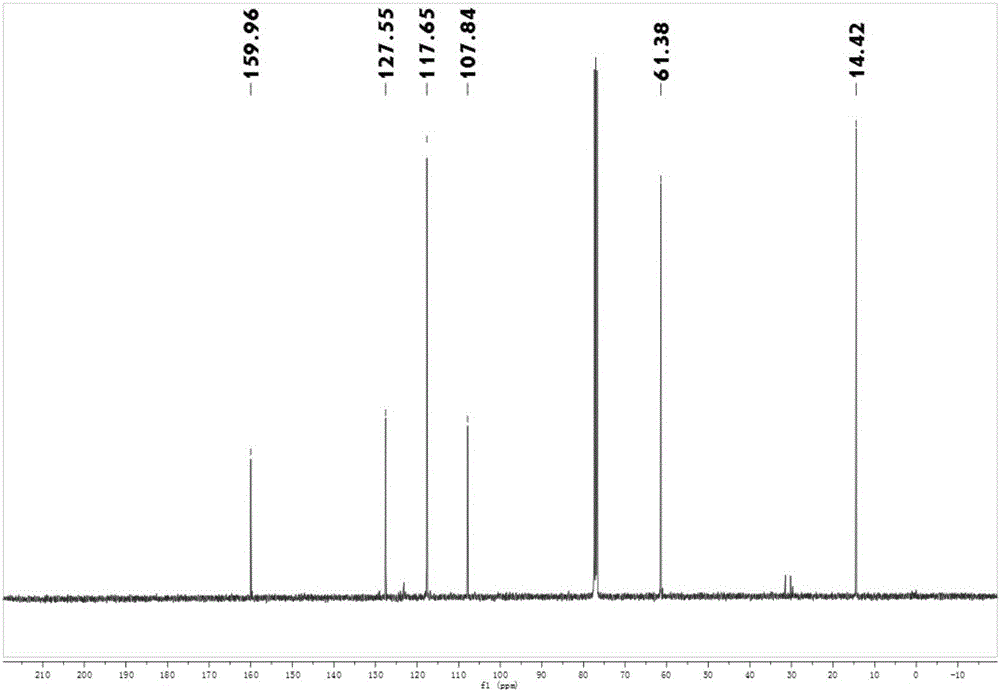
所承认的,并且包括适于将本发明化合物给予哺乳动物的药学上可接受的材料、组合物或载剂。载体包括参与将药剂从一个器官或身体一个部分带到或输送到另一器官或身体另一部分的液体或固体填充剂、稀释剂、赋形剂、溶剂或包封材料。各载体必须在与制剂的其它成分相容且对患者无害方面是“可接受的”。可以用作药学上可接受载体的材料的一些例子包括:糖类,如乳糖、葡萄糖和蔗糖;淀粉类,如玉米淀粉和马铃薯淀粉;纤维素、及其衍生物,例如羧甲基纤维素钠、乙基纤维素、和醋酸纤维素;粉状西黄蓍胶;麦芽;明胶;滑石;赋形剂,如可可脂和栓剂蜡类;油类,如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;二醇类,如丙二醇;多元醇类,如甘油、山梨糖醇、甘露醇和聚乙二醇;酯类,如油酸乙酯和月桂酸乙酯;琼脂;缓冲剂,如氢氧化镁和氢氧化铝;海藻酸;无热原水;等渗盐水;林格氏溶液;乙醇;磷酸盐缓冲溶液;药物制剂中使用的其它无毒的相容性物质。湿润剂、乳化剂和润滑剂(如十二烷基硫酸钠和硬脂酸镁)、以及着色剂、脱模剂、包衣剂、甜味剂、矫味剂和芳香剂、防腐剂和抗氧化剂也可以存在于这些组合物中。本发明的制剂包括适于口服给药、经鼻给药、局部给药、口腔给药、舌下给药、直肠给药、阴道给药和/或胃肠外给药的制剂。这些制剂可以方便地采用单位剂型的形式并且可以利用药学领域中众所周知的任何方法制备。以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于限制本发明的范围。实施例中采用的实施条件可以根据具体厂家的条件做进一步调整,未注明的实施条件通常为常规实验中的条件。一、化合物4的合成,实施例1化合物4的合成,其包括如下步骤:(1)向三颈烧瓶中加入45ml无水乙醚和吡咯(5.003g,75mmol),并向其中缓慢滴加130ml无水乙醚和三氯乙酰氯(9.5ml,85mmol)的混合液,2h内滴完,继续搅拌回流1h,然后用饱和碳酸钠溶液淬灭反应,收集有机相,用无水硫酸镁干燥,活性炭脱色,减压蒸除溶剂后得到白色粉末状固体2-三氯乙酰基吡咯20.43g,产率94.25%。1HNMR(300MHz,CDCl3)δ9.72(brs,1H),7.40(s,1H,-CH-),7.18(s,1H,-CH-),6.37-6.40(m,1H,-CH-);(2)向500ml的三颈烧瓶中加入2-三氯乙酰基吡咯(25.10g,119mmol)和90ml乙腈,控制温度为-15℃,然后向三颈烧瓶中慢慢加入NBS(21.06g,119mmol),TLC跟踪点板,约3h后停止反应。出现大量白色不溶物,抽滤,滤饼用水淋洗。向滤液中加入200ml冰水后,出现大量紫灰色固体,抽滤,将得到的滤饼干燥,并用乙醇和水混合溶剂重结晶得到白色固体4-溴-2-三氯乙酰基吡咯32.94g,产率95.8%。1HNMR(300MHz,CDCl3)δ9.70(brs,1H),7.42(d,1H,J=0.9Hz,-CH-),7.17(d,1H,J=1.8Hz,-CH-);(3)向250ml的三颈烧瓶中加入金属钠(0.69g,30mmol)和20ml乙醇,待金属钠完全溶解后,烧瓶内温度降至10℃,取化合物4-溴-2-三氯乙酰基吡咯(16.28g,56mmol)和40ml无水乙醇,溶解后缓慢滴加到三颈烧瓶内,室温搅拌,TLC跟踪点板,约30min后反应结束。减压蒸除溶剂,然后向所得的残渣中加入冰乙醇和水(体积比为2:1)混合溶剂30ml,搅拌,抽滤得橘红色固体4-溴-1H-吡咯-2-甲酸乙酯10.71g,产率87.7%。1HNMR(300MHz,CDCl3)δ9.51(brs,1H),6.94(s,1H,-CH-),6.89(s,1H,-CH-),4.32(q,2H,J=7.2Hz,-CH2-),1.35(t,3H,J=7.2Hz,-CH3)(4)向50ml三颈烧瓶中分别加入DMF(0.662g,9.065mmol),POCl3(1.1917g,7.762mmol),20ml二氯甲烷,室温搅拌10min,在加入4-溴-1H-吡咯-2-甲酸乙酯(0.565g,2.592mmol),继续室温反应24h,TLC点板反应完全,再向反应液中加入80ml水,再用CH2Cl2(3×30ml)萃取,饱和食盐水洗涤,无水硫酸镁干燥,旋干,柱层析分离,得到5-醛基-4-溴-1H-吡咯-2-甲酸乙酯0.5866g,产率92%。1HNMR(400MHz,CDCl3)δ9.83(s,1H),9.72(s,1H),6.96(d,J=2.7Hz,1H),4.38(q,J=7.1Hz,2H),1.38(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ179.59,159.67,130.72,128.14,117.77,107.96,61.86,14.24;二、以化合物4为基础原料合成各种5-取代-4-溴-1H-吡咯-2-甲酸酯类化合物实施例2化合物5的合成向50ml三口烧瓶中加入4-溴-1H-吡咯-2-甲酸乙酯(0.695g,3.2mmol),倒入20ml乙腈,使其完全溶解,再加入NCS(0.427g,3.2mmol),室温下搅拌24h,TLC跟踪点板。反应结束后,将反应液旋干,柱层析分离,得到5-氯-4-溴-1H-吡咯-2-甲酸乙酯0.5g,产率:62%。1HNMR(400MHz,CDCl3)δ10.26(s,1H),6.89(d,J=2.9Hz,1H),4.36(q,J=7.1Hz,1H),1.37(t,J=7.1Hz,1H);13CNMR(101MHz,CDCl3)δ160.46,121.63,120.67,117.74,96.54,61.34,14.33,化合物6的合成向50ml三颈烧瓶中加入4-溴-1H-吡咯-2-甲酸乙酯(0.68g,3.2mmol),倒入10ml二氯甲烷使其完全溶解,再加入I2(0.812g,3.2mmol),三氟乙酸银(0.22g,1mmol)。通入N2保护,反应避光,室温搅拌。TLC跟踪点板,约7h后反应停止。加入Na2SO3溶液淬灭,饱和食盐水洗涤,乙酸乙酯萃取(3×25ml),旋干,柱层析分离,得到产物5-碘-4-溴-1H-吡咯-2-甲酸乙酯0.7724g,产率81%。1HNMR(400MHz,CDCl3)δ10.20(s,1H),6.88(d,J=2.8Hz,1H),4.38(q,J=7.1Hz,2H),1.37(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ159.96,127.55,117.65,107.84,76.90,61.38,14.42,化合物7的合成向50ml三颈烧瓶中,10mlDMF,原料4(99.2mg,0.4mmol),FeSO4·7H2O(834.06mg,3mmol),再加入三氟甲磺酰氯(85ul,0.8mmol),逐滴加入35%H2O2(100ul),N2保护,冰浴搅拌15min,再加入FeSO4·7H2O(834.06mg,3mmol),再加入三氟甲磺酰氯(42.5ul,0.4mmol),逐滴加入35%H2O2(50ul),继续冰浴搅拌15min,TLC跟踪点板。待反应完全,旋干,柱层析分离,得产物55mg,产率51%。1HNMR(400MHz,CDCl3)δ10.30(s,1H),6.93(d,J=1.7Hz,1H),4.38(q,J=7.1Hz,2H),1.38(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ160.37,124.44,122.55(q,J=39.0Hz),119.96(q,J=268.9Hz),118.27,98.19(q,J=2.7Hz),61.78,14.18,化合物8的合成冰浴下,将4-溴-1H-吡咯-2-甲酸乙酯(0.654g,3.mmol)溶于10ml乙腈溶液中,再逐滴加入氯磺酰异氰酸酯(0.27ml,3mmol),转移至室温反应17h,TLC跟踪点板,直至反应完全。再加入DMF(1.17ml)淬灭,50℃继续加热2h,冷却至室温,再反应2h,将混合物倒在40ml冰水上,3×25ml乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸镁干燥。旋干,柱层析分离,得到产物5-氰基-4-溴-1H-吡咯-2-甲酸乙酯0.4374g,产率60%。1HNMR(400MHz,CDCl3)δ11.04(s,1H),6.94(d,J=2.4Hz,1H),4.45(q,J=8.6,3.3Hz,2H),1.40(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ159.92,127.02,117.20,111.34,108.01,107.04,62.58,14.23,三、以化合物9为原料进一步合成5-取代-4-溴-1H-吡咯-2-甲酸酯类化合物实施例3化合物10的合成向50ml三颈烧瓶中,加入DAST(0.124g,0.6mmol)的二氯甲烷溶液(20ml),再加入原料9(0.124g,0.5mmol),室温搅拌过夜,TLC跟踪点板。待反应完全,加入H2O(25ml),EA萃取(3×25ml),饱和盐水洗涤,无水硫酸镁干燥,旋干,柱层析分离,得产物0.113g,产率85%。1HNMR(400MHz,CDCl3)δ9.90(s,1H),6.90–6.88(m,1H),6.72(t,J=53.7Hz,1H),4.37(q,J=7.1Hz,2H),1.37(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ160.08,126.05(t,J=24.7Hz),124.60,116.89,108.57(t,J=235.4Hz),98.85(t,J=7.0Hz),61.45,14.29。化合物11的合成向50ml三颈烧瓶中先加入5-醛基-4-溴-1H-吡咯-2-甲酸乙酯(65.4mg,0.3mmol),4-氯苯胺(51mg。0.4mmol),无水硫酸镁(0.5g)乙醚20ml,室温搅拌24h,TLC跟踪点板。待反应完全后,过滤,旋干,加入甲醇20ml使其完全溶解,再加入硼氢化钠(37.84mg,1mmol),室温反应10min,TLC点板。旋干,柱层析分离,得到5-(4-氯-N-甲基苯胺)-4-溴-1H-吡咯-2-甲酸乙酯0.104g,产率97%。1HNMR(400MHz,CDCl3)δ9.62(s,1H),7.12(d,J=8.7Hz,2H),6.88(d,J=2.6Hz,1H),6.54(d,J=8.7Hz,2H),4.30(s,2H),4.29–4.22(q,2H),1.31(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ160.70,145.80,132.36,129.26,123.56,122.09,117.47,114.51,96.30,60.84,40.58,14.33。化合物12的合成向50ml三颈烧瓶中加入5-醛基-4-溴-1H-吡咯-2-甲酸乙酯(0.1796g,0.73mmol),硼氢化钠(0.11g,2.91mmol)甲醇20ml,N2保护,剧烈搅拌,室温反应2h,TLC点板确认反应完全,柱层析分离,得到产物5-苄醇-4-溴-1H-吡咯-2-甲酸乙酯0.1652g,产率92%。1HNMR(400MHz,CDCl3)δ9.65(s,1H),6.86(d,J=2.5Hz,1H),4.72(s,2H),4.32(q,J=7.1Hz,2H),1.36(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ160.66,133.67,122.30,117.21,95.78,60.85,56.71,14.36。化合物13的合成向50ml三颈烧瓶中先加入5-苄醇-4-溴-1H-吡咯-2-甲酸乙酯(100mg,0.4mmol),二氯甲烷20ml使其完全溶解,冰浴,滴入乙酰氯(154.59mg,1mmol),再加入三乙胺(1.2mmol,120mg),室温搅拌6h,TLC点板。待反应完全,旋干,加入碳酸钾溶液(50ml),乙酸乙酯萃取(3×25ml),无水硫酸镁干燥,旋干,柱层析分离,得到产品5-(乙酸甲酯)-4-溴-1H-吡咯-2-甲酸乙酯0.1g,产率87%。1HNMR(400MHz,CDCl3)δ9.49(s,1H),6.84(d,J=2.8Hz,1H),5.08(s,2H),4.32(q,J=7.1Hz,2H),2.11(s,3H),1.35(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ171.99,160.05,129.06,123.50,116.64,99.54,60.88,56.94,20.82,14.36。化合物14的合成向50ml三颈烧瓶中先加入5-苄醇-4-溴-1H-吡咯-2-甲酸乙酯(100mg,0.4mmol),二氯甲烷20ml使其完全溶解,冰浴,滴入对甲基苯甲酰氯(154.59mg,1mmol),再加入三乙胺(1.2mmol,120mg),撤去冰浴,室温搅拌6h,TLC点板。待反应完全,旋干,加入碳酸钾溶液(50ml),乙酸乙酯萃取(3×25ml),无水硫酸镁干燥,旋干,柱层析分离,得到产品5-(对甲基苯甲酸甲酯)-4-溴-1H-吡咯-2-甲酸乙酯0.1g,产率71%。1HNMR(400MHz,CDCl3)δ9.79(s,1H),7.93(d,J=8.1Hz,2H),7.23(d,J=8.1Hz,2H),6.85(d,J=2.7Hz,1H),5.31(s,2H),4.31(q,J=7.1Hz,2H),2.40(s,3H),1.33(t,J=7.1Hz,3H),1.33(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ167.54,160.14,144.31,129.93,129.31,129.19,126.59,123.53,116.67,99.69,60.87,57.23,21.73,14.36。化合物15的合成向50ml三颈烧瓶中加入5-醛基-4-溴-1H-吡咯-2-甲酸乙酯(60mg,0.243mmo),再加入10mlDMSO使其完全溶解,冰浴,再将配好的磷酸氢二钠溶液(34.5mg,0.243mmol),H2O(5ml)和亚氯酸钠溶液NaClO2(90.44mg,1mmol),H2O(5ml)分别滴入和,撤去冰浴,室温搅拌24h,TLC点板。待反应完全后,调PH至2,加入H2O(40ml),用乙酸乙酯(3×25ml)萃取,无水硫酸镁干燥,过滤,旋干,干燥,得到白色固体5-羧基-4-溴-1H-吡咯-2-甲酸乙酯80mg,产率96%。1HNMR(400MHz,DMSO)δ13.24(s,1H),12.81(s,1H),6.90(s,1H),4.25(q,J=7.1Hz,2H),1.29(t,J=7.1Hz,3H);13CNMR(101MHz,DMSO)δ159.73,158.46,125.04,123.95,117.67,101.25,60.03,13.54。化合物16的合成向50ml三颈烧瓶中先加入5-羧酸-4-溴-1H-吡咯-2-甲酸乙酯(94.5mg,0.378mmol),再加入乙醇20ml,使其完全溶解,冰浴,滴入氯化亚砜0.115ml,85℃回流一夜,TLC点板。待反应完全,旋干,加入碳酸钾溶液(50ml),乙酸乙酯萃取(3×25ml),无水硫酸镁干燥,旋干,柱层析分离,得到产物5-(甲酸乙酯)-4-溴-1H-吡咯-2-甲酸乙酯0.103g,产率94%。1HNMR(400MHz,CDCl3)δ9.81(s,1H),6.93(d,J=2.8Hz,1H),4.38(dq,J=14.1,7.1Hz,4H),1.39(dt,J=13.9,7.1Hz,6H);13CNMR(101MHz,CDCl3)δ159.60,159.34,125.42,123.35,118.74,103.41,61.45,14.28。化合物17的合成向50ml三颈烧瓶中先加入5-羧酸-4-溴-1H-吡咯-2-甲酸乙酯(125mg,0.5mmol),二氯甲烷20ml,使其完全溶解,冰浴,滴入草酰氯(0.55mol,0.465ml),滴入2滴DMF撤出冰浴,室温搅拌2h,直至没有气泡生成,旋干,冰浴,加入苯酚(0.5mmol,44mg),DMAP(6mg,0.05mmol),三乙胺(151.785mg,1.5mmol),二氯甲烷20ml,室温搅拌6h,TLC点板。待反应完全后,旋干,加入碳酸钾溶液(50ml),乙酸乙酯萃取(3×25ml),无水硫酸镁干燥,旋干,柱层析分离,得到产物5-(甲酸苯酯)-4-溴-1H-吡咯-2-甲酸乙酯0.1764g,产率88%。1HNMR(400MHz,CDCl3)δ10.02(s,1H),7.43(dd,J=10.8,5.0Hz,2H),7.28(dd,J=7.5Hz,1H),7.25–7.20(m,2H),6.99(d,J=5.6Hz,1H),4.38(q,J=7.1Hz,2H),1.38(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ159.58,157.61,149.97,129.56,126.41,126.26,122.59,121.54,119.00,104.95,61.67,14.27。化合物(18)的合成向50ml三颈烧瓶中先加入5-羧酸-4-溴-1H-吡咯-2-甲酸乙酯(125mg,0.5mmol),二氯甲烷20ml,使其完全溶解,冰浴,滴入草酰氯(0.55mol,0.465ml),滴入2滴DMF撤出冰浴,室温搅拌2h,直至没有气泡生成,旋干,冰浴,加入4-氯苯胺(0.5mmol,63.5mg),DMAP(6mg,0.05mmol),三乙胺(151.785mg,1.5mmol),二氯甲烷20ml,室温搅拌6h,TLC点板。待反应完全后,旋干,加入碳酸钾溶液(50ml),乙酸乙酯萃取(3×25ml),无水硫酸镁干燥,旋干,柱层析分离,得到产物5-[N-(4-氯苯基)甲酰胺]-4-溴-1H-吡咯-2-甲酸乙酯0.172g,产率93%。1HNMR(400MHz,CDCl3)δ10.16(s,1H),8.74(s,1H),7.59(d,J=8.8Hz,2H),7.34(d,J=8.8Hz,2H),6.97(d,J=3.0Hz,1H),4.37(q,J=7.1Hz,2H),1.38(t,J=7.1Hz,3H);13CNMR(101MHz,CDCl3)δ159.32,156.68,135.67,130.02,129.25,125.87,125.25,121.44,118.25,97.46,61.47,14.30。上述实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人是能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1