一种栀子酰胺A-他克林二联体化合物及其制法和应用的制作方法
本发明属于药物化学领域,特别涉及一种栀子酰胺A-他克林二联体化合物及其制法和在制备治疗神经退行性疾病药物中的应用。
背景技术:
神经退行性疾病是通过毁坏维持正常大脑功能的神经元,引起脑损失或躯体功能障碍的一类疾病。临床上主要有阿茨海默症(Alzheimer’s disease,AD)、肌肉萎缩性侧索硬化症、共济失调毛细血管扩张症、牛海绵状脑症、克雅二氏症、亨廷顿症、小脑萎缩症、多发性硬化症、帕金森症(Parkinson’s disease,PD)、原发性侧索硬化及脊椎性肌萎缩症等,其中,最为常见、最多发的是AD和PD。神经退行性疾病的病人群主要是老年人,随着人口老年化加剧,发病率将逐年增加,近年来也发现发病人群年轻化的趋势。
PD是以运动减少,肌肉强直和震颤为特征的神经退行性疾病,其病理特征为黑质和纹状体多巴胺(dopamine,DA)神经元变性减少,纹状体多巴胺递质、细胞内嗜酸性路易斯小体形成。PD的药物治疗可以分成抗胆碱能药、金刚烷胺、多巴胺替代疗法、多巴胺受体激动剂、单胺氧化酶-B抑制剂、COMT抑制剂六大类。一般来讲,治疗后临床症状有一定的好转,但并不能改善多巴胺能神经元功能和延缓退化,随着服药时间的延长和给药剂量的增加,效果降低,不良反应增大。
AD是原发性神经退行性疾病,病变部位主要发生在大脑,是一组异质性多病机制的疾病,其成因涉及到多个方面,主要与脑内胆碱水平的降低、β-淀粉样蛋白(Aβ)的聚集或沉淀、氧化应激、钙离子水平失调和金属离子水平异常增高以及中枢神经炎症等多种因素相关。临床表现为记忆功能障碍、智力减退和行为人格退化等症状。老年痴呆的发病率呈逐年上升趋势,临床上用于改善AD的药物主要是抗胆碱能和NMDA受体抑制剂类等,至今仍缺少理想的治疗药物,对AD的发病机制的研究和新治疗药物的开发仍有待深入的研究。
中医药在涉及退行性疾病中的临床应用具有悠久的历史,在治疗和预防方面有独特的优势。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种栀子酰胺A-他克林二联体化合物。
本发明另一目的在于提供上述栀子酰胺A-他克林二联体化合物的制备方法。
本发明再一目的在于提供上述栀子酰胺A-他克林二联体化合物在制备治疗神经退行性疾病药物中的应用,尤其是在制备治疗老年痴呆症药物中的应用。
本发明的目的通过下述方案实现:
一种栀子酰胺A-他克林二联体化合物,其结构式如式I所示:
其中,n为1~7的正整数。
式I中,n=1时,式I化合物命名为TNG-1;
n=2时,式I化合物命名为TNG-2;
n=3时,式I化合物命名为TNG-3;
n=4时,式I化合物命名为TNG-4;
n=5时,式I化合物命名为TNG-5;
n=6时,式I化合物命名为TNG-6;
n=7时,式I化合物命名为TNG-7。
优选的,上述的栀子酰胺A-他克林二联体化合物为TNG-1、TNG-2、TNG-4、TNG-5。
更优选的,上述的栀子酰胺A-他克林二联体化合物为TNG-1、TNG-5。
上述的栀子酰胺A-他克林二联体化合物的制备方法,具体包括以下步骤:
(1)以京尼平为原料,首先将京尼平C-1位上的半缩醛转化为内酯,再将内酯转化为内酰胺,最后将C-10位羟基氧化成醛,即得到中间体化合物DG;
(2)以邻氨基苯甲酸甲酯为原料,首先通过水解、环化反应制备得到氯代他克林,然后将其与对应的二胺NH2-CH2-(CH2)n-NH2通过亲核取代反应制备得到中间体化合物TN-n;
(3)将制备得到的中间体化合物DG和中间体化合物TN-n通过胺化还原反应得到栀子酰胺A-他克林二联体化合物。
以n=1为例,其对应使用的二胺为乙二胺。
所述的栀子酰胺A-他克林二联体化合物的制备路线如下式所示:
优选的,上述的栀子酰胺A-他克林二联体化合物的制备方法,具体包括以下步骤:
(1)中间体化合物G-2的制备:取京尼平和缚酸剂溶于溶剂,室温搅拌5~10min后,滴加溶于相同溶剂中的1.0~5.0倍于京尼平摩尔量的羟基保护试剂,然后室温下搅拌反应1.5~3.0小时,反应完毕后将所得反应液纯化即得中间体化合物G-2;
(2)中间体化合物G-3的制备:取中间体化合物G-2溶于溶剂,加入1.0~5.0倍于中间体G-2摩尔量的氧化剂,室温下搅拌反应2~8h,然后将所得反应液纯化即得中间体G-3;
(3)中间体化合物GA的制备:取中间体化合物G-3和乙酸胺溶于溶剂,在油浴中加温至110℃反应,搅拌反应2小时后,除去溶剂,然后向残留物加入重蒸的四氢呋喃溶解,再加入三氟乙酸,80℃下继续搅拌反应2小时,将所得的反应液纯化即得中间体化合物GA;
(4)中间体化合物DG的制备:取中间体化合物GA溶于溶剂,加入1.0~5.0倍于中间体化合物GA摩尔量的氧化剂,室温下搅拌反应2~8h,然后将所得反应液纯化即得中间体化合物DG;
(5)邻氨基苯甲酸的制备:称取邻氨基苯甲酸甲酯,加入氢氧化钠水溶液,搅拌回流反应2~18小时;反应完毕,冷却至室温,用稀盐酸调溶液pH至3~4,然后滴加冰醋酸,抽滤,滤饼水洗,真空干燥即得邻氨基苯甲酸;
(6)中间体化合物T-3的制备:取邻氨基苯甲酸和环己酮,用甲苯溶解,然后回流反应5h,反应完毕将所得反应液纯化即得中间体T-3;
(7)氯代他克林的制备:取中间体T-3加入到三氯氧磷中,回流反应2h,反应结束后将所得反应液纯化即得氯代他克林;
(8)中间体化合物TN-n(n=1-7)的制备:取氯代他克林、相应二胺和催化量的碘化钾溶于正戊醇溶液中,回流反应8h,然后将所得反应液纯化即得中间体化合物TN-n(n=1-7);
(9)栀子酰胺A-他克林二联体化合物的制备(n=1-4):取中间体化合物DG和中间体化合物TN-n溶于溶剂,加入催化量经活化的分子筛,在室温搅拌1~6h后,冰浴条件下预冷5min,加入1~6倍于中间体化合物DG摩尔量的还原剂,冰浴下反应2~16h,反应完毕后将所得反应液纯化即得栀子酰胺A-他克林二联体化合物;
(10)栀子酰胺A-他克林二联体化合物的制备(n=5-7):取中间体化合物DG和中间体化合物TN-n溶于溶剂,加入催化量经活化的分子筛,在室温搅拌1~6h后,加入1~6倍于中间体化合物DG摩尔量的还原剂,室温反应2~8h,反应完毕后将所得反应液纯化即得栀子酰胺A-他克林二联体化合物(n=5-7)。
步骤(1)中所述的溶剂为四氢呋喃(THF)、二氯甲烷(DCM)、N,N-二甲基甲酰胺(DMF)中的至少一种,优选为四氢呋喃;所述的缚酸剂为咪唑、吡啶、三乙基胺中的至少一种,优选为咪唑;所述的羟基保护剂为二甲基叔丁基氯化硅、三甲基氯化硅、苄溴中的至少一种,优选为二甲基叔丁基氯化硅;所述的纯化是指将所得反应液直接旋干溶剂,然后将所得的残留物经柱层析分离即得纯化后的中间体化合物G-2,柱层析分离采用的洗脱剂优选为体积比为10:1的石油醚和乙酸乙酯的混合物。
步骤(1)中所用的京尼平和缚酸剂的摩尔比为1.0:(1.1~3.0)。
步骤(2)中所述的溶剂为二氯甲烷、THF、甲苯中的至少一种,优选为二氯甲烷;所述的氧化剂为戴斯-马丁试剂、二氧化锰、PCC中的至少一种,优选为戴斯-马丁试剂;所述的纯化是指向所得反应液中加入饱和碳酸氢钠溶液和饱和硫代硫酸钠溶液淬灭反应,然后用二氯甲烷萃取,合并有机相,用饱和氯化钠溶液洗涤后,再用无水硫酸钠干燥,过滤后用旋转蒸发仪除去溶剂,残留物用柱层析分离即得中间体G-3,柱层析分离所用的洗脱剂优选为体积比为25:1的石油醚和乙酸乙酯的混合物。
步骤(3)中所述的溶剂为吡啶、甲苯、氯苯中的至少一种,优选为吡啶;所述的纯化指向反应液中加入饱和碳酸氢钠溶液中和过量的三氟乙酸,直至不再有气体冒出,然后用二氯甲烷(DCM)萃取,合并有机相,饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤后用旋转蒸发仪除溶剂,残留液用柱层析分离纯化即得纯化后的GA,柱层析所使用的洗脱剂优选为体积比为1:1的石油醚和乙酸乙酯的混合物;
步骤(3)中所用的中间体化合物G-3和乙酸胺的摩尔比为1.0:(1.5~5.0)。
步骤(4)中所述的溶剂为N,N-二甲基甲酰胺(DMF)、四氢呋喃(THF)、二氯甲烷(DCM)中的至少一种,优选为二氯甲烷;所述的氧化剂为PCC、戴斯-马丁氧化试剂或Jone’s试剂中的至少一种,优选为戴斯-马丁氧化试剂;所述的纯化指将所得反应液冰浴冷却后过滤,并用二氯甲烷洗涤滤渣,合并滤液,然后将所得滤液除去溶剂,将所得残留物经柱层析分离即得纯化后的中间体化合物DG,柱层析所使用的洗脱剂优选为体积比为1:1的石油醚和乙酸乙酯的混合物。
步骤(6)中所述的纯化是指将所得反应液冷却至室温,再冰浴继续冷却,然后抽滤,将所得滤饼分别用甲苯洗、乙醇洗,真空干燥即得纯化后的中间体T-3。
步骤(6)中所用的邻氨基苯甲酸和环己酮的摩尔比为1.0:(1.2-3.0)。
步骤(7)中所述的纯化是指将所得反应液冷却至室温后,将反应液倒入冰水中,用饱和氢氧化钠溶液调节pH值到11,DCM萃取,萃取液用饱和氯化钠溶液洗涤,无水硫酸钠干燥,除去溶剂后直接烘干处理即得纯化后的氯代他克林。
步骤(7)中所用的中间体T-3和三氯氧磷的摩尔比为1.0:(1.0-30.0)。
步骤(8)中TN-1对应的二胺为乙二胺,TN-2对应的二胺为1,3-丙二胺,TN-3对应的二胺为1,4-丁二胺,TN-4对应的二胺为1,5-戊二胺,TN-5对应的二胺为1,6-己二胺,TN-6对应的二胺为1,7-庚二胺,TN-7对应的二胺为1,8-辛二胺;所述的纯化是指将所得反应液除去溶剂,然后加入氢氧化钠溶液,氯仿萃取,萃取液用饱和氯化钠溶液洗涤,无水硫酸钠干燥,再除去溶剂,残留物经柱层析分离即得纯化后的中间体化合物TN-n(n=1-7),柱层析所用的洗脱剂为体积比为25:1:0.13的氯仿、甲醇、氨水的混合液。
步骤(8)中所用的二胺的量为氯代他克林的摩尔量的6倍。
步骤(9)中所述的溶剂为四氢呋喃、甲醇、二氯甲烷、二甲基亚砜(DMSO)、1,2-二氯乙烷中的至少一种,优选为甲醇;所述的还原剂为硼氢化钠、氰基硼氢化钠、三乙酰氧基硼氢化钠中的至少一种,优选为氰基硼氢化钠;所述的纯化是指将所得反应液蒸干溶剂,然后用饱和氯化铵淬灭,乙酸乙酯萃取,合并萃取液,饱和氯化钠溶液洗涤,无水硫酸钠干燥,再除去溶剂,残留物经色谱分离即得栀子酰胺A-他克林二联体化合物(n=1-4);所述的色谱分离是指将残留物用甲醇溶解,然后过ODS柱,利用高效液相色谱仪分离纯化(洗脱剂,甲醇:水=65:35,V/V)。
步骤(10)中所述的溶剂为四氢呋喃、甲醇、二氯甲烷、DMSO、1,2-二氯乙烷中的至少一种,优选为1,2-二氯乙烷;所述的还原剂为硼氢化钠、氰基硼氢化钠、三乙酰氧基硼氢化钠中的至少一种,优选为三乙酰氧基硼氢化钠;所述的纯化是指将所得反应液蒸干溶剂,然后用饱和碳酸氢钠淬灭,乙酸乙酯萃取,合并萃取液,饱和氯化钠溶液洗涤,无水硫酸钠干燥,再除去溶剂,残留物经色谱分离即得栀子酰胺A-他克林二联体化合物(n=5-7)。
所述的色谱分离优选为将残留物用甲醇溶解,然后过ODS柱,利用高效液相色谱仪分离纯化(洗脱剂,甲醇:水=45:55,V/V)。
上述的栀子酰胺A-他克林二联体化合物可用于制备治疗神经退行性疾病的药物,尤其可用于制备治疗老年痴呆症的药物;尤其可用于制备治疗多巴能神经元损伤所导致的老年痴呆症药物。
本发明相对于现有技术,具有如下的优点及有益效果:
本发明的化合物为首次报道,其对神经退行性疾病有治疗效果,可用于制备治疗神经退行性疾病的药物。相比于其它现有技术,本发明的化合物制备工艺简单、疗效优于他克林。
附图说明
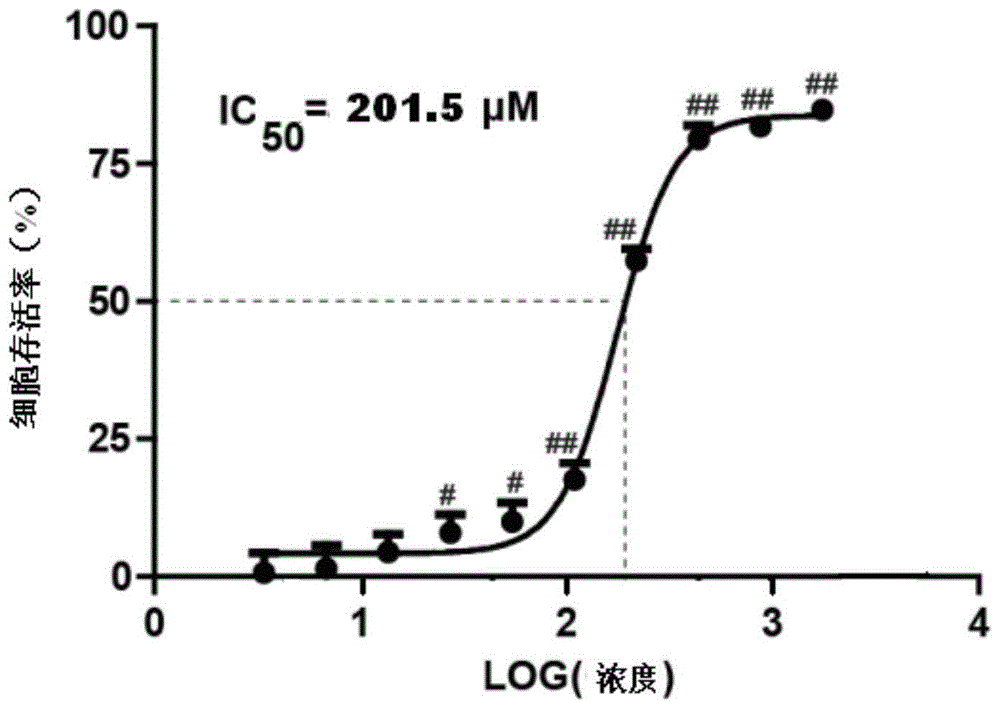
图1为实施例8中6-OHDA对PC12细胞的半抑制浓度结果图。
图2为实施例8中TNG-1~TNG-7及阳性对照药对6-OHDA诱导的神经损伤保护作用图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例中所用试剂如无特殊说明均可从市场上常规购得。
实施例1:10-N-(2-(10-氨基代栀子酰胺A)氨基)乙基他克林(TNG-1)的合成
包括以下步骤:
(1)中间体化合物G-2的制备:称取京尼平(340.2mg,1.5mmol)和咪唑(308.5mg,4.5mmol)于25mL的圆底烧瓶内,用3mL干燥的四氢呋喃溶液溶解;然后称取二甲基叔丁基氯化硅(TBSCl)(408.8mg,2.7mmol),用干燥的四氢呋喃溶液溶解,将其滴加到反应体系中。室温下搅拌反应1.5小时。待反应完毕,直接旋干溶剂。残留液柱层析分离纯化(石油醚:乙酸乙酯=10:1(V:V)),得白色化合物451.2mg,收率89%。白色化合物的表征数据为1H NMR(300MHz,CDCl3)δ:7.55(s,1H,3-H),5.83(brs,1H,1-H),4.80(d,J=12Hz,1H,CH),4.31-4.25(q,J=12,18Hz,2H),3.70(s,3H,-OCH3),3.31(brs,1H,CH),3.18-3.09(q,J=9,18Hz,1H,CH),2.85-2.67(m,1H,CH),2.46-2.41(m,1H,CH),2.05-1.97(m,1H,H),0.93(s,9H,-SiC(CH3)3),0.09(s,3H,-SiCH3),0.08(s,3H,-SiCH3);13C NMR(75MHz,CDCl3)δ:172.4,156.8,148.1,130.0,114.2,100.2,65.7,54.2,50.8,42.4,40.1,29.0,21.8,2.7,2.6;ESI-MS(m/z):363.2[M+Na]+。上述数据证实该化合物为中间体化合物G-2。
(2)中间体化合物G-3的制备:称取中间体化合物G-2(261.1mg,0.77mmol)于25mL的圆底烧瓶中,加入5mL干燥的二氯甲烷溶液溶解,称取戴斯-马丁试剂(491.3mg,1.15mmol)分批加入到上述反应体系中,反应体系呈混悬液状态,室温下搅拌反应4h。待反应完毕,向反应体系中加入10mL饱和碳酸氢钠溶液和10mL饱和硫代硫酸钠溶液淬灭反应,然后用二氯甲烷萃取3次(3×50mL),合并有机相,用饱和氯化钠溶液洗涤后,再用无水硫酸钠干燥,过滤后用旋转蒸发仪除去溶剂。残留物用柱层析分离纯化(石油醚:乙酸乙酯=25:1(V:V)),得白色化合物208.2mg,收率80%。1H NMR(300MHz,CDCl3)δ:7.49(s,1H),5.87-5.86(m,1H),4.52-4.40(m,2H),3.78(s,3H,-OCH3),3.65(d,J=8.1Hz,1H),3.56-3.47(m,1H),2.96-2.88(m,1H),2.27-2.17(m,1H),0.91(s,9H,-SiC(CH3)3),0.09(s,3H,-SiCH3),0.09(s,3H,-SiCH3);13C NMR(75MHz,CDCl3)δ:166.5,166.1,148.4,141.0,127.5,113.3,61.4,51.9,45.9,39.3,36.2,25.9,18.4,5.4,5.4;ESI-MS(m/z):337.41[M-H]-。上述数据证实该化合物为中间体G-3。
(3)中间体化合物GA的制备:称取中间体化合物G-3(68.1mg,0.2mmol)和乙酸铵(32.3mg,0.41mmol)置于耐压管中,并加入2mL干燥的吡啶溶解,在油浴中加温至110℃反应,搅拌反应2小时后,此时反应液呈黑色,于60-75℃减压条件下将吡啶蒸干。随后将残留物直接转移至耐压管中,用重蒸的四氢呋喃溶液溶解,加入0.2mL三氟乙酸,将油浴锅降温至80℃,继续搅拌反应2小时。待反应完毕,向反应体系中加入饱和碳酸氢钠溶液中和过量的三氟乙酸,直至不再有气体冒出,用二氯甲烷萃取3次(3×50mL),合并有机相,饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤后用旋转蒸发仪除溶剂。残留液用柱层析分离纯化(石油醚:乙酸乙酯=1:1(V:V)),得淡黄色粉末固体15.1mg,收率33.8%。1H NMR(300MHz,CDCl3)δ:7.75(s,1H,NH),7.19(s,1H),5.85(s,1H),4.40-4.30(q,J=9.6Hz,2H),3.76(s,3H,-OCH3),3.68(d,J=8.1Hz,1H),3.61-3.53(m,1H),2.97-2.91(m,1H),2.28-2.22(m,1H);13C NMR(75MHz,CDCl3)δ:172.3,166.8,140.5,132.6,129.5,111.2,60.9,51.6,49.4,40.1,37.2;ESI-MS(m/z):222.3[M-H]-。上述数据证实该化合物为中间体化合物GA。
(4)中间体化合物DG的制备:称取中间体化合物GA(100.4mg,0.45mmol)于25mL的圆底烧瓶中,加入8mL干燥的二氯甲烷溶液溶解,称取戴斯-马丁试剂(384.5mg,0.90mmol)分批加入到上述反应体系中,反应体系呈混悬液状态,室温下搅拌反应6h。反应完毕,冰浴下放置0.5h,然后直接过滤,并用二氯甲烷洗涤,合并滤液,蒸干溶剂。残留物用柱层析分离纯化(石油醚:乙酸乙酯=1:1(V:V)),得白色化合物84.5mg,收率85%。产物的分析数据如下:1H NMR(300MHz,CDCl3)δ:9.84(s,1H,-CHO),8.59(s,1H),7.26(m,1H),6.97(s,1H),3.94(d,J=9.0Hz,1H),3.73(s,3H,-OCH3),3.53(m,1H),3.11(m,1H),2.39(m,1H);13C NMR(75MHz,CDCl3)δ:188.2,170.1,166.7,150.8,144.9,134.2,109.0,51.7,44.7,40.3,37.5;ESI-MS(m/z):220.1[M-H]-。上述数据证实该化合物为中间体化合物DG。
(5)邻氨基苯甲酸的制备:称取邻氨基苯甲酸甲酯(12.1g,80mmol)于250mL的圆底烧瓶中,加入50mL的饱和氢氧化钠水溶液,在105℃油浴中加热反应2.5h后。反应完毕,冷却至室温,此时溶液呈橙黄色,用1.0mol/L稀盐酸调体系pH值至3-4左右,此时有少量白色固体析出;向反应体系中滴加冰醋酸,有大量白色固体析出,抽滤,滤饼水洗,真空干燥得白色固体10.4g,收率95%。熔点144-146℃(文献值:144-146℃)(Carlier P.R.,Han Y.F.,Chow E.S.Bioorg.Med.Chem.1999,7,351-357)。上述数据证实该化合物为邻氨基苯甲酸。
(6)中间体化合物T-3的制备:称取邻氨基苯甲酸(10.0g,73.0mmol)和环己酮(8.6g,87.6mmol)于250mL的圆底烧瓶中,加入35mL甲苯溶解,接分水器,在120℃油浴中加热反应5h后。反应完毕,溶液呈深棕色。冷却至室温的过程中有少量白色针状结晶析出,继续使用冰浴使其冷却至0℃,有大量棕黄色固体析出,然后进行抽滤,滤饼分别用甲苯洗、乙醇洗,真空干燥得黄色固体16.04g,收率89%。熔点142-144℃(文献值:143-145℃)(Carlier P.R.,Han Y.F.,Chow E.S.Bioorg.Med.Chem.1999,7,351-357)。上述数据证实该化合物为中间体化合物T-3。
(7)中间体化合物T-4的制备:量取70mL三氯氧磷于500mL圆底烧瓶中,往体系中分批加入中间体化合物T-3(13.9g,64.2mmol),在120℃油浴中加热反应2h,反应完毕,反应液呈橙红色。冷却至室温后,将反应液缓慢倒入冰水中,用饱和氢氧化钠溶液调节pH值至11左右,此时反应液由橙红色澄清溶液变为淡黄色浑浊液,并有部分固体析出,二氯甲烷萃取3次(3×100mL),合并有机相,用饱和氯化钠溶液洗涤后,无水硫酸钠干燥,旋转蒸除溶剂,直接烘干处理,得淡黄色片状固体13.82g,收率91%。熔点65-67℃(文献值:66.5-67.7℃)(Carlier P.R.,Han Y.F.,Chow E.S.Bioorg.Med.Chem.1999,7,351-357)。上述数据证实该化合物为中间体化合物T-4。
(8)中间体化合物TN-1的制备:取中间体化合物T-4(0.25g,1.15mmol)、乙二胺(8.65mmol)和催化量的碘化钾加入到25mL的圆底烧瓶中,量取5mL正戊醇加入反应体系,在160℃油浴中加热回流8h,此时反应液呈棕黄色油状液,反应毕,用真空泵泵在油浴65-80℃条件下蒸除正戊醇,然后加入20mL 2mol/L的氢氧化钠溶液,用氯仿萃取3次(3×30mL),合并有机相,用饱和氯化钠溶液洗涤后,再用无水硫酸钠干燥,旋转蒸发仪除溶剂,残留物用柱层析分离纯化(氯仿:甲醇:氨水=25:1:0.13,V/V/V),得黄色油状液0.20g,收率72%。1H NMR(300MHz,CD3OD)δ:8.11-8.08(m,1H),7.79-7.76(m,1H),7.58-7.52(m,1H),7.40-7.34(m,1H),3.73-3.68(m,2H),3.63-3.59(m,2H),2.98(t,J=12Hz,2H),2.79(t,J=12Hz,2H),1.93-1.89(m,4H)。上述数据证实该化合物为中间体化合物TN-1。
(9)10-N-(2-(10-氨基代栀子酰胺A)氨基)乙基他克林(TNG-1)的制备:称取中间体化合物DG(0.25mmol)于25mL的圆底烧瓶中,用10mL干燥甲醇溶解,加入中间体化合物TN-1(0.25mmol),然后再加入少量的经活化分子筛,在室温搅拌4h后,移至0℃的恒温反应浴中,稳定5min,称取氰基硼氢化钠(0.05g,0.75mmol)加入到反应体系中,继续搅拌过夜。反应毕,蒸干溶剂,饱和氯化铵淬灭,用乙酸乙酯萃取3次(3×20mL),合并有机相,用饱和氯化钠溶液洗涤后,再用无水硫酸钠干燥,旋转蒸发仪除溶剂,残留物用适量甲醇溶解,过ODS柱,高效液相制备(甲醇:水=65:35,V/V),得到目标化合物48.4mg,收率约为45%。1H NMR(300MHz,CD3OD)δ:8.11-8.08(d,J=6Hz,1H),7.78-7.75(d,J=6Hz,1H),7.58-7.52(m,1H),7.39-7.34(m,1H),7.14(s,1H),5.69(s,1H),3.72(s,3H,-OCH3),3.70-3.60(m,2H),3.58-3.44(m,2H),3.31-3.29(m,2H)2.96(t,J=12Hz,2H),2.87-2.80(m,1H),2.83-2.78(m,2H),2.80-2.72(m,2H),2.18-2.02(m,1H),1.93-1.82(m,4H);13C NMR(75MHz,CD3OD)δ:172.2,168.8,159.0,153.1,147.6,141.7,136.5,134.8,129.9,129.1,127.8,124.4,121.3,117.3,111.0,52.0,49.9,49.6,49.4,48.5,41.2,38.3,34.0,26.1,24.0,23.6;ESI-MS(m/z):447.5[M+H]+,446.0[M-H]-.上述数据证实该化合物为10-N-(2-(10-氨基代栀子酰胺A)氨基)乙基他克林(TNG-1)。结构如下所示:
实施例2:10-N-(3-(10-氨基代栀子酰胺A)氨基)正丙基他克林(TNG-2)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-2的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,3-丙二胺((8.65mmol)。得黄色油状液0.25g,收率约为85%。1H NMR(300MHz,CDCl3)δ:8.16-8.13(d,J=9Hz,1H),7.88-7.85(d,J=9Hz,1H),7.64-7.59(m,1H),7.44-7.38(m,1H),4.1(s,2H,-NH2),2.66(t,J=12Hz,2H),2.39(t,J=12Hz,2H),2.27-2.19(m,2H),1.55-1.42(m,4H)。上述数据证实该化合物为中间体化合物TN-2;
(4)10-N-(3-(10-氨基代栀子酰胺A)氨基)正丙基他克林(TNG-2)的制备:按实施例1步骤(9)制备。将TN-1(0.25mmol)替换为TN-2(0.25mmol)。得淡黄色油状液体46.6mg,收率为42%。1H NMR(300MHz,CD3OD)δ:8.12-8.09(m,1H),7.76-7.73(m,1H),7.58-7.52(m,1H),7.39-7.34(m,1H),7.15(s,1H),5.68(s,1H),3.72(s,3H,-OCH3),3.65-3.61(t,J=12Hz,2H),3.46(m,2H),3.42-3.37(m,2H),2.96(t,J=12Hz,2H),2.84-2.78(m,1H),2.73(t,J=12Hz,2H),2.64-2.60(m,2H),2.14-2.06(m,1H),1.94-1.78(m,6H);13C NMR(75MHz,CD3OD)δ:170.9,167.4,157.6,151.7,146.3,139.9,133.4,128.4,127.8,126.4,123.4,123.0,119.7,115.2,109.6,50.6,48.5,46.5,46.1,39.8,37.0,36.8,32.7,30.2,22.7,22.3;ESI-MS(m/z):461.5[M+H]+,460.1[M-H]-。以上数据证明化合物为10-N-(3-(10-氨基代栀子酰胺A)氨基)正丙基他克林(TNG-2),结构如下所示:
实施例3:10-N-(4-(10-氨基代栀子酰胺A)氨基)正丁基他克林(TNG-3)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-3的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,4-丁二胺(8.65mmol)。得黄色油状液体0.25g,收率约为81%。1H NMR(300MHz,CDCl3)δ:7.94-7.92(d,J=6.0Hz,1H),7.88-7.86(d,J=6.0Hz,1H),7.53-7.49(m,1H),7.32-7.29(m,1H),4.18(s,1H,-NH-),3.47(t,J=12.0Hz,2H),3.01(m,2H),2.94(m,4H),2.71-2.66(m,4H),1.67-.62(m,2H),1.54-1.49(m,2H)。上述数据证实该化合物为中间体化合物TN-3。
(4)10-N-(4-(10-氨基代栀子酰胺A)氨基)正丁基他克林(TNG-3)的制备:按实施例1步骤(9)制备。将TN-1(0.25mmol)替换为TN-3(0.25mmol)。得淡黄色油状液体43.5mg,收率38%。1H NMR(300MHz,CD3OD)δ:8.10-8.07(m,1H),7.77-7.74(m,1H),7.56-7.51(m,1H),7.37-7.32(m,1H),7.15(s,1H),5.70(s,1H),3.72(s,3H,-OCH3),3.56-3.52(t,J=12Hz,2H),3.47-3.38(m,3H),2.96(t,J=12Hz,2H),2.86-2.78(m,1H),2.74(t,J=12Hz,2H),2.56-2.51(m,2H),2.17-2.09(m,1H),1.94-1.85(m,5H),1.71-1.50(m,4H);13C NMR(75MHz,CD3OD)δ:172.3,168.8,159.2,153.1,147.9,141.2,134.8,129.7,129.3,128.0,124.7,124.4,121.3,116.9,110.0,52.0,50.0,49.8,49.5(2),41.2,38.4,34.2,30.0,27.6,26.2,24.1,23.7;ESI-MS(m/z):475.4[M+H]+,474.2[M-H]-。以上数据证明化合物为10-N-(4-(10-氨基代栀子酰胺A)氨基)正丁基他克林(TNG-3),结构如下所示:
实施例4:10-N-(5-(10-氨基代栀子酰胺A)氨基)正戊基他克林(TNG-4)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-4的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,5-戊二胺(8.65mmol)。得黄色油状液体0.29g,收率约为89%。1H NMR(300MHz,CD3OD)δ:8.06-8.02(m,1H),7.76-7.72(m,1H),7.55-7.50(m,1H),7.34-7.28(m,1H),3.49(t,J=12Hz,2H),2.98(t,J=12Hz,2H),2.71(t,J=12Hz,2H),2.53(t,J=15Hz,2H),1.91-1.86(m,4H),1.65-1.56(m,2H),1.44-1.24(m,4H)。上述数据证实该化合物为中间体化合物TN-4。
(4)10-N-(5-(10-氨基代栀子酰胺A)氨基)正戊基他克林(TNG-4)的制备:按实施例1步骤(9)。将TN-1(0.25mmol)替换为TN-4(0.25mmol)。得淡黄色油状液体50.8mg,收率43%。1H NMR(300MHz,CD3OD)δ:8.10-8.08(m,1H),7.77-7.74(m,1H),7.57-7.51(m,1H),7.38-7.32(m,1H),7.16(s,1H),5.74(s,1H),3.72(s,3H,-OCH3),3.56-3.51(t,J=12Hz,2H),3.48-3.43(m,3H),2.96(t,J=12Hz,2H),2.89-2.80(m,1H),2.74(t,J=12Hz,2H),2.58-2.45(m,2H),2.21-2.11(m,1H),1.94-1.86(m,5H),1.70-1.60(m,2H),1.56-1.46(m,2H),1.42-1.35(m,2H);13C NMR(75MHz,CD3OD)δ:172.3,168.8,159.2,153.2,147.9,141.1,134.8,129.7,129.3,128.0,124.7,124.4,121.3,116.8,110.0,52.0,50.1,49.9,49.7,41.3,38.4,38.3,34.2,32.1,29.9,26.2,25.6,24.1,23.7;ESI-MS(m/z):475.4[M+H]+,474.2[M-H]-。以上数据证明化合物为10-N-(5-(10-氨基代栀子酰胺A)氨基)正戊基他克林(TNG-4),结构如下所示:
实施例5:10-N-(6-(10-氨基代栀子酰胺A)氨基)正己基他克林(TNG-5)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-5的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,6-己二胺(8.65mmol)。得黄色油状液体0.23g,收率67%。1H NMR(300MHz,CD3OD)δ:8.07-8.03(m,1H),7.78-7.74(m,1H),7.54-7.49(m,1H),7.35-7.29(m,1H),3.47(t,J=12Hz,2H),2.95(t,J=12Hz,2H),2.69(t,J=12Hz,2H),2.54(t,J=15Hz,2H),1.90-1.85(m,4H),1.64-1.55(m,2H),1.43-1.23(m,6H);13C NMR(75MHz,CD3OD)δ:159.3,153.3,148.2,129.8,128.3,124.8,124.5,121.5,116.9,48.3,42.5,34.4,33.8,32.4,28.0,27.9,26.3,24.3,23.9。上述数据证实该化合物为中间体化合物TN-5;
(4)10-N-(6-(10-氨基代栀子酰胺A)氨基)正己基他克林(TNG-5)的制备:称取中间体化合物DG(0.25mmol)于25mL圆底烧瓶中,用10mL 1,2-二氯乙烷溶解,加入中间体化合物TN-5(0.25mmol),再加入少量经活化后的分子筛,室温搅拌2h后,分批加入三乙酰氧基硼氢化钠(0.16g,0.75mmol)到反应体系中,继续在室温条件下搅拌反应4小时。反应完毕,蒸干溶剂,饱和碳酸氢钠溶液淬灭,乙酸乙酯萃取3次(3×20mL),合并有机相,饱和氯化钠溶液洗涤,无水硫酸钠干燥,旋蒸除去溶剂,残留物用适量甲醇溶解,过ODS柱,高效液相制备(洗脱剂,甲醇:水=45:55,V/V),得淡黄色油状液体44.9mg,收率37%。1H NMR(300MHz,CD3OD)δ:8.10-8.07(m,1H),7.77-7.74(m,1H),7.56-7.51(m,1H),7.37-7.32(m,1H),7.15(s,1H),5.75(s,1H),3.71(s,3H,-OCH3),3.54-3.49(t,J=15Hz,2H),3.48-3.41(m,3H),2.96(t,J=12Hz,2H),2.89-2.80(m,1H),2.73(t,J=12Hz,2H),2.57-2.44(m,2H),2.19-2.10(m,1H),1.94-1.85(m,4H),1.68-1.58(m,2H),1.53-1.43(m,2H),1.40-1.26(m,4H);13C NMR(75MHz,CD3OD)δ:172.3,168.8,159.1,153.2,147.9,141.0,134.8,129.7,129.4,128.0,124.7,124.4,121.3,116.8,110.0,52.0,50.1,49.8,49.6(2),41.3,38.4,38.3,34.2,32.2,30.1,28.0,27.8,26.2,24.1,23.7;ESI-MS(m/z):503.5[M+H]+,502.9[M-H]-。以上数据证明化合物为10-N-(6-(10-氨基代栀子酰胺A)氨基)正己基他克林(TNG-5),结构如下所示:
实施例6:10-N-(7-(10-氨基代栀子酰胺A)氨基)正庚基他克林(TNG-6)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-6的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,7-庚二胺(8.65mmol)。得黄色油状液体0.26g,收率71%。1HNMR(300MHz,CD3OD)δ:8.08-8.05(d,J=9Hz,1H),7.77-7.74(d,J=9Hz,1H),7.56-7.50(m,1H),7.36-7.31(m,1H),3.49(t,J=15Hz,2H),2.96(t,J=12Hz,2H),2.71(t,J=12Hz,2H),2.60(t,J=12Hz,2H),1.89-1.87(m,4H),1.62-1.55(m,2H),1.43-1.39(m,2H),1.25(m,6H)。上述数据证实该化合物为中间体化合物TN-6;
(4)10-N-(7-(10-氨基代栀子酰胺A)氨基)正庚基他克林(TNG-6)的制备:按实施例5步骤(4)制备。将TN-5(0.25mmol)替换为TN-6(0.25mmol)。得淡黄色油状液体67.5mg,收率54%。1H NMR(300MHz,CD3OD)δ:8.11-8.08(m,0.6Hz,1H),7.78-7.75(m,1H),7.58-7.53(m,1H),7.39-7.34(m,1H),7.17(s,1H),5.79(s,1H),3.73(s,3H,-OCH3),3.56-3.51(t,J=15Hz,2H),3.51-3.44(m,3H),2.98(t,J=12Hz,2H),2.91-2.82(m,1H),2.75(t,J=12Hz,2H),2.59-2.48(m,2H),2.21-2.12(m,1H),1.96-1.87(m,4H),1.69-1.60(m,2H),1.53-1.44(m,2H),1.40-1.29(m,6H);13C NMR(75MHz,CD3OD)δ:172.4,168.8,159.0,153.4,147.8,140.8,134.9,129.8,129.7,127.8,124.7,124.5,121.3,116.7,110.1,52.0,49.9,49.7,49.6,41.3,38.4,38.3,34.1,32.2,30.0,28.2,27.8,26.2,24.1,23.7;ESI-MS(m/z):517.8[M+H]+,515.4[M-H]-。以上数据证明化合物为10-N-(7-(10-氨基代栀子酰胺A)氨基)正庚基他克林(TNG-6),结构如下所示:
实施例7:10-N-(8-(10-氨基代栀子酰胺A)氨基)正辛基他克林(TNG-7)的合成
包括以下步骤:
(1)中间体化合物DG的制备:按实施例1步骤(1)至(4)制备;
(2)中间体化合物T-4的制备:按实施例1步骤(5)至(7)制备;
(3)中间体化合物TN-7的制备:按实施例1步骤(8)制备。将乙二胺(8.65mmol)替换为1,8-辛二胺(8.65mmol)。得黄色油状液体0.24g,收率65%。1HNMR(300MHz,CD3OD)δ:8.08-8.05(d,J=9Hz,1H),7.77-7.74(d,J=9Hz,1H),7.56-7.50(m,1H),7.36-7.31(m,1H),3.49(t,J=15Hz,2H),2.96(t,J=12Hz,2H),2.71(t,J=12Hz,2H),2.60(t,J=12Hz,2H),1.89-1.87(m,4H),1.62-1.55(m,2H),1.43-1.39(m,2H),1.25(m,8H)。上述数据证实该化合物为中间体化合物TN-7;
(4)10-N-(8-(10-氨基代栀子酰胺A)氨基)正辛基他克林(TNG-7)的制备:按实施例5步骤(4)制备。将TN-5(0.25mmol)替换为TN-7(0.25mmol)。得淡黄色油状液体66.8mg,收率约为52%。1H NMR(300MHz,CD3OD)δ:8.08-8.06(m,1H),7.77-7.74(m,1H),7.56-7.50(m,1H),7.36-7.31(m,1H),7.16(s,1H),5.76(s,1H),3.71(s,3H,-OCH3),3.55-3.42(m,5H),2.96(t,J=12Hz,2H),2.89-2.80(m,1H),2.71(t,J=12Hz,2H),2.56-2.46(m,2H),2.20-2.11(m,1H),1.94-1.86(m,5H),1.65-1.56(m,2H),1.51-1.42(m,2H),1.34-1.18(m,8H);13C NMR(75MHz,CD3OD)δ:172.3,168.8,159.1,153.2,147.9,141.0,134.8,129.7,129.4,128.0,124.7,124.5,121.3,116.7,110.0,52.0,49.9(2),49.7,41.3,38.4,38.3,34.2,32.3,30.4,30.3,30.2,28.2,27.8,26.2,24.1,23.7;ESI-MS(m/z):531.8[M+H]+.以上数据证明化合物为10-N-(8-(10-氨基代栀子酰胺A)氨基)正辛基他克林(TNG-7),结构如下所示:
实施例8:化合物TNG-1~TNG-7对6-羟多巴(6-OHDA)诱导的PC12神经细胞损伤的保护作用
本实施例中,(褐家鼠)肾上腺嗜铬细胞瘤PC12细胞购自中国科学院上海细胞库(上海,中国);高糖培养基(DMEM)购自Invitrogen公司(美国);胎牛血清(FBS)购自Biological Industries公司(以色列);胰蛋白酶购自美国Gibco公司;磷酸盐缓冲液(PBS)购自武汉博士德公司;噻唑蓝(MTT)购自美国Sigma公司。
实验步骤:将对数期生长的PC12细胞经过胰酶消化后,离心,用完全培养基(含10%FBS的DMEM)制成细胞悬液,计数,调整细胞浓度为6×104/mL。将细胞接种于96孔细胞培养板,100μL/孔,将96孔板置于37℃,5%CO2的培养箱中培养24h,待细胞贴壁后,弃去旧培养基,加入不同浓度的药物。每孔加入14μL的MTT(5mg/mL,溶剂为PBS),于细胞培养箱中孵育2~4h;弃上层液体,每孔加入150μL DMSO,振摇10min,使板底部的紫色结晶充分溶解;使用酶标仪(Thermo MK3,USA)检测570nm出的吸光度值,记录结果,计算细胞增长抑制率,每次实验至少重复三次。同时设置正常组和空白对照组,正常组为含细胞但不加待试药物的测试孔;空白对照组为只含有DMSO的测试孔;计算公式:细胞存活率(%)=(药物组-空白对照组)/(正常组-空白对照组)×100%;
将PC12细胞按1×104/孔接种于96孔板中,于培养箱中孵育24h后,弃去旧培养基,分别加入不同浓度的6-羟多巴(6-OHDA)处理24h,同时设置正常组和空白对照组,其中正常组为含细胞不加6-OHDA的测试孔,空白对照组为只含DMSO的测试孔;细胞存活率(%)=[1-(6-OHDA组-空白对照组)/(正常组-空白对照组)]×100%,结果如图1所示,从图1中可以看出,6-OHDA的半抑制浓度IC50=201.5μM。因此,选择200μM的6-OHDA作为评估待测药物神经保护作用的浓度。
将PC12细胞按1×104/孔接种于96孔板中,37℃、5%CO2及饱和湿度条件下培养24h后,换液为不含血清的DMEM培养基,200μM 6-OHDA预先处理2小时后加入终浓度为3、10、30μM的TNG-1~TNG-7以及阳性对照药Tacrine和栀子酰胺GA。处理24h,随后加入10μL MTT(10mg/mL),继续培养4h后去上清液,每孔加入150μL DMSO,微振荡后,用全自动酶标仪在570nm波长处读取吸光度(A)值。同时设置正常组和空白对照组,正常组为含细胞不经6-OHDA处理不加待试药物的测试孔,空白对照组为只加DMSO的测试孔;按以下公试计算细胞存活率:
细胞存活率(%)=(药物组-空白对照组)/(正常组-空白对照组)×100%
其中,药物组为含细胞经6-OHDA处理并加入待试药物的测试孔;空白对照组为只加DMSO的测试孔;正常组为含细胞不经6-OHDA处理不加待试药物的测试孔。结果如图2所示,从图2中可以看到,TNG-1和TNG-5在30μM呈现最佳的神经保护作用;TNG-2和TNG-4分别在10μM和30μM显示弱的的神经损伤保护作用;TNG-3、TNG-6和TNG-7的在所检测的浓度范围内均未显示出神经保护作用;阳性药他克林(Tacrine)于30μM下显示弱的神经损伤保护作用;栀子酰胺A(GA)于30μM左右显示一定的保护作用,但比TNG-1和TNG-5弱。联合给药(栀子酰胺A:他克林=1:1)实验组均未呈现明显的神经毒性保护作用。
可见,化合物TNG-1,TNG-4,TNG-5在30μM浓度下具有明显的神经损伤保护作用。初步说明二联体的协同效果。
因此,上述的栀子酰胺A-他克林二联体化合物可用于制备治疗神经退行性疾病的药物,尤其可用于制备治疗老年痴呆症的药物;尤其可用于制备治疗多巴能神经元损伤所导致的老年痴呆症药物。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!