一种盐酸溴己新的制备方法与流程
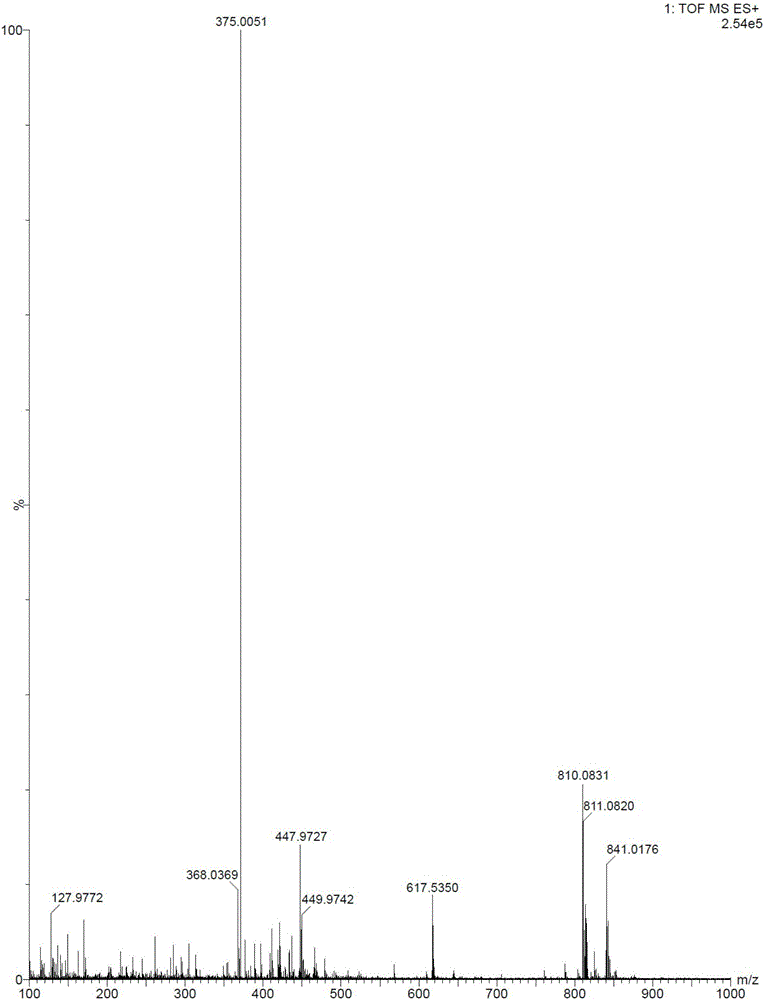
本发明属于药物合成领域,具体涉及一种盐酸溴己新的制备方法。
背景技术:
:盐酸溴己新(BromhexineHydrochloride,N-(2-氨基-3,5-二溴苄基)-N-甲基环己胺盐酸盐)是由勃林格殷格翰公司研制的药物,临床上用于急慢性支气管炎、哮喘、支气管扩张、肺气肿的治疗,尤其适用于白色粘痰咳出的困难者,及因痰液广泛阻塞小支气管引起的气急等症。目前,关于盐酸溴己新的合成工艺文献报道比较多,而综合文献分析发现,报道的盐酸溴己新的合成方法包括以下几种,其合成路线如下:路线一路线二路线三路线一的工艺中涉及的缩合剂大多为二环己基碳二亚胺(DCC),其副产物二环己基脲(DCU)毒性大,去除难度较大,其残留风险影响盐酸溴己新的用药安全。路线二的反应类型均较为简单,但工艺步骤长,操作繁琐,生产周期长;而且工艺过程中涉及安全风险较大的卤代试剂,对反应设备的要求较高;同时,卤代试剂活性较高,可能导致苯环上的溴原子被取代而产生副反应,影响产品质量。虽然路线三的工艺较为简便:醋酸催化条件下,加热、甲苯分水进行缩合反应,缩短了生产周期。但是该路线的甲苯分水步骤需要高温反应,度反应设备要求高;同时还存在反应不完全的风险,工业化生产中存在安全风险。综上,提供一种盐酸溴己新的制备方法,工艺简单,操作简便,反应条件温和,反应收率较好,有较好的工业化基础成为了本领域技术人员亟待解决的问题。技术实现要素:本发明提供了一种盐酸溴己新的制备方法,该制备方法工艺简单,操作简便,反应条件温和,收率好,纯度高,杂质含量少,所需原料易得,具有良好的工业化基础。本发明所述的一种盐酸溴己新的制备方法,包括如下步骤:a:2-氨基-3,5-二溴苯甲醛、N-甲基环己胺与钛酸酯在溶剂中反应生成过渡态;b:还原剂原位还原所述过渡态,生成溴己新游离碱;c:所述溴己新游离碱成盐制备盐酸溴己新;反应式为:优选地,所述R为C1-C6的直链烷基或支链烷基。进一步优选地,所述直链烷基为甲基、乙基、正丙基、正丁基,所述支链烷基为异丙基、异丁基。优选地,所述步骤a中,所述2-氨基-3,5-二溴苯甲醛与所述N-甲基环己胺的摩尔比为1:0.8~1:4,所述2-氨基-3,5-二溴苯甲醛与所述钛酸酯的摩尔比为1:1~1:3。进一步优选地,所述2-氨基-3,5-二溴苯甲醛与所述N-甲基环己胺的摩尔比为1:2~1:3。优选地,所述步骤a中,所述溶剂选自醇类溶剂、醚类溶剂、卤代烃类溶剂、腈类溶剂和亚砜类溶剂中的一种或几种。进一步优选地,所述溶剂选自为甲醇、乙醇、异丙醇、正丁醇、四氢呋喃、1,4-二氧六环、二氯甲烷中的一种或多种。优选地,所述步骤b中,所述还原剂选自硼氢化钠、硼氢化钾、硼氢化锂中的一种或几种,所述2-氨基-3,5-二溴苯甲醛与所述还原剂的摩尔比为1:0.3~1:3,所述步骤a、所述步骤b、所述步骤c的反应温度为-10~50℃。进一步优选地,所述步骤a、所述步骤b、所述步骤c的反应温度为20~30℃。与现有技术相比,本发明的有益效果:本发明以2-氨基-3,5-二溴苯甲醛和N-甲基环己胺为原料,经还原胺化、成盐制备盐酸溴己新。采用本发明制备盐酸溴己新,缩短了盐酸溴己新的反应步骤,且所需原料易得。本发明中,2-氨基-3,5-二溴苯甲醛结构中的醛基在-10~50℃的温度下能与N-甲基环己胺和钛酸酯发生反应产生含有缩醛结构的过渡态,所得过渡态无需分离纯化,可直接被还原剂还原生成溴己新游离碱。该工艺可在同一反应容器中进行,避免了现有技术中需对中间产物进行分离或纯化的步骤。本发明工艺简单,操作简便,反应条件温和,所需原料易得,有利于工业化生产。采用本发明制备盐酸溴己新收率好,纯度高,杂质含量少,能够保证药品的安全、有效。附图说明图1为盐酸溴己新的高分辨质谱图。图2为盐酸溴己新的核磁共振氢谱图。具体实施方式以下具体实施方式可以对本发明的内容做出更为详细的说明,但本发明的主题范围不局限于以下具体实施例,凡是基于本
发明内容所实现的技术、工艺均属于本发明的范围。实施例1本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基环己胺溶于20ml甲醇中,室温搅拌条件下将4g钛酸四异丙酯缓慢滴入到反应液中,加完后继续室温搅拌反应5h;将0.3g硼氢化钠分批加入反应液中,室温搅拌反应2h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。室温、搅拌条件下,向上述有机相中滴入5ml6N盐酸,搅拌析晶3h。过滤得一白色固体,乙醇重结晶、干燥,得到2.3g盐酸溴己新,收率:78%。HRMS-ESI(m/z):375.0051(M+H+);1H-NMR(400MHz,CD3OD)δ=7.70(d,1H),7.48(d,1H),4.50(d,1H),4.23(d,1H),3.40~3.34(m,1H),2.70(s,3H),2.18~2.09(m,2H),1.97(s,2H),1.76~1.57(m,3H),1.47~1.37(m,2H),1.27~1.24(m,1H)ppm.实施例2本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、4gN-甲基环己胺溶于20ml甲醇中,于10℃搅拌条件下将2g钛酸四异丙酯缓慢滴入到反应液中,加完后继续于10℃搅拌反应5h;将0.3g硼氢化钠分批加入反应液中,室温搅拌反应2h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。于10℃搅拌条件下,向上述有机相中滴入10ml6N盐酸,搅拌析晶3h。过滤得一白色固体,乙醇重结晶、干燥,得到2.1g盐酸溴己新,收率:71%。实施例3本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基环己胺溶于20ml异丙醇中,于30℃搅拌条件下将4g钛酸四异丙酯缓慢滴入到反应液中,加完后继续室温搅拌反应5h;将0.3g硼氢化钠分批加入反应液中,于30℃搅拌反应2h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。于30℃搅拌条件下,向上述有机相中滴入5ml6N盐酸,搅拌析晶3h。过滤得一白色固体,乙醇重结晶、干燥,得到2.4g盐酸溴己新,收率:81%。实施例4本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基环己胺溶于20ml甲醇中,于0℃搅拌条件下将4.9g钛酸四丁酯缓慢滴入到反应液中,加完后继续于0℃搅拌反应6h;将0.3g硼氢化钠分批加入反应液中,室温搅拌反应2h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。于0℃搅拌条件下,向上述有机相中滴入5ml6N盐酸,搅拌析晶3h。过滤得白色固体,乙醇重结晶、干燥,得到2.0g盐酸溴己新,收率:68%。实施例5本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、2gN-甲基环己胺溶于20ml1,4-二氧六环中,于-10℃搅拌条件下将4g钛酸四异丙酯缓慢滴入到反应液中,加完后继续于-10℃搅拌反应8h;将0.3g硼氢化钠分批加入反应液中,室温搅拌反应4h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。于-10℃搅拌条件下,向上述有机相中滴入5ml6N盐酸,搅拌析晶3h。过滤得白色固体,乙醇重结晶、干燥,得到2.3g盐酸溴己新,收率:79%。实施例6本实施例中的原料均来源于市售。将2.0g2-氨基-3,5-二溴苯甲醛、1.2gN-甲基环己胺溶于20ml甲醇中,于50℃搅拌条件下将4g钛酸四异丙酯缓慢滴入到反应液中,加完后继续于50℃搅拌反应5h;将0.5g硼氢化钾分批加入反应液中,室温搅拌反应2h。TLC检测反应完全。反应结束后,向反应液中加入20ml水淬灭反应,析出大量固体,过滤,滤饼用乙酸乙酯洗涤3次,合并滤液和洗液,乙酸乙酯萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。于50℃搅拌条件下,向上述有机相中滴入5ml6N盐酸,搅拌析晶3h。过滤得白色固体,乙醇重结晶、干燥,得到2.2g盐酸溴己新,收率:74%。实施例7对比例:采用
背景技术:
中路线一方法制备盐酸溴己新。将11g2-氨基-3,5-二溴苯甲酸、2g4-二甲胺基吡啶和5gN-羟基丁二酰亚胺(HOSu)溶于20mL二氯甲烷中,搅拌条件下将溶有10g二环己基碳二亚胺(DCC)的二氯甲烷溶液缓慢滴入上述反应液中,继续搅拌反应,析出大量固体,继续反应2h。将18gN-甲基环己胺加入反应液中,搅拌反应2h。TLC监控反应完全。向反应液中加入10mL6NHCl淬灭反应,过滤,萃取得有机相。搅拌条件下,向上述有机相中分批加入1.2gLiBH4,加完后,加热至40℃反应。TLC监控反应完全。反应结束后,向反应液中加入20ml水淬灭反应,二氯甲烷萃取得有机相,依次用碳酸钠溶液、饱和食盐水洗涤有机相。搅拌条件下,向上述有机相中滴入20ml6N盐酸,搅拌析晶3h。过滤得白色固体,乙醇重结晶,得到9.5g盐酸溴己新,收率:62%。实施例8对比例:采用
背景技术:
中路线二的方法制备盐酸溴己新。11.2g2-氨基-3,5-二溴苯甲醛、20ml无水乙醇,搅拌下分批次加入0.95g、NaBH4,控制温度在10~40℃,非均相反应,TLC监控反应完全。反应完毕后,向反应体系中加入50ml水,搅拌下滴加2N盐酸调溶液pH=5~6,继续搅拌30min,将固体过滤,滤饼用水(20ml×3)洗涤,干燥,得还原产物为白色固体。将上述还原产物分批加入到30ml氯化亚砜中,滴加过程中保持温度0~40℃,滴加完毕后常温下搅拌反应,反应时间3~4小时。反应完毕后,将过量的氯化亚砜常温下减压浓缩,残余物中加入20ml石油醚,搅拌20min,过滤,石油醚(10ml×3)洗涤,30℃下减压干燥,得氯带中间体为黄色固体。将上述氯带中间体分批加入到11.34gN-甲基环己胺中,再加入20ml无水乙醇,常温下搅拌反应。TLC监控反应进程,反应时间3~4小时。反应完毕后,将反应液中的溶剂减压浓缩,向残余物中加入40ml乙酸乙酯,有固体析出,继续搅拌20min,将固体过滤,滤液用HCl/EA溶液跳pH=5~6,降温至0~5℃下搅拌析晶,过滤,干燥,得到盐酸溴己新粗品,甲醇重结晶得到盐酸溴己新约6.8g,收率:41%。实施例9对比例:采用
背景技术:
中路线三的方法制备盐酸溴己新。依次将110gN-甲基环己胺、90g2-氨基-3,5-二溴苯甲醇加入到反应瓶中,搅拌条件下向反应液中加入50g冰醋酸和60mL甲苯,加热分水(内温约110~120℃,外温约130~140℃),TLC监控反应进度至原料不再减少。减压浓缩后,加入120mL乙醇溶解,搅拌条件下滴加6NHCl至溶液体系pH为2,冷却析晶过夜,过滤得到盐酸溴己新粗品,精制得盐酸溴己新约93g,收率:70%实施例10盐酸溴己新中杂质的测定。取实施例1-9所得盐酸溴己新,测定杂质含量。实验方法:照《中国药典》(2015年版,二部)盐酸溴己新【检查】项下有关物质测定方法测定,结果见下表。表1杂质含量测定表最大单一杂质(%)总杂质(%)实施例10.0210.033实施例20.0370.058实施例30.0320.069实施例40.0430.062实施例50.0200.044实施例60.0410.072实施例7(对比例)0.1150.215实施例8(对比例)0.0760.144实施例9(对比例)0.0920.185由表1可知,本发明的最大单一杂质和杂质总量都优于工艺路线一、工艺路线二和工艺路线三。综上,采用本发明所得的盐酸溴己新的收率更高,杂质含量更少。且本发明工艺简单,操作简便,反应条件温和,所需原料易得,具有良好的工业化基础。上述实施例仅为本发明的优选实施方式之一,不应当用于限制本发明的保护范围,但凡在本发明的主体设计思想和精神上作出的毫无实质意义的改动或润色,其所解决的技术问题仍然与本发明一致的,均应当包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1