用于评估杂合性丢失的方法与材料与流程
本申请是申请号为201280070358.0、申请日为2012年12月21日、发明名称为“用于评估杂合性丢失的方法与材料”的中国发明专利申请的分案申请,原申请为国际申请号为pct/us2012/071380的国家阶段申请,该国际申请要求申请日分别为2011年12月21日和2012年6月1日,申请号分别为61/578,713和61/654,402的美国申请的优先权。相关申请的交叉引用本申请请求在2011年12月21日提交的序列号为61/578,713的美国临时专利申请和2012年6月1日提交的序列号为61/654,402的美国临时申请之优先权,其内容在此通过引用的方式全文并入本文。背景1.
技术领域:
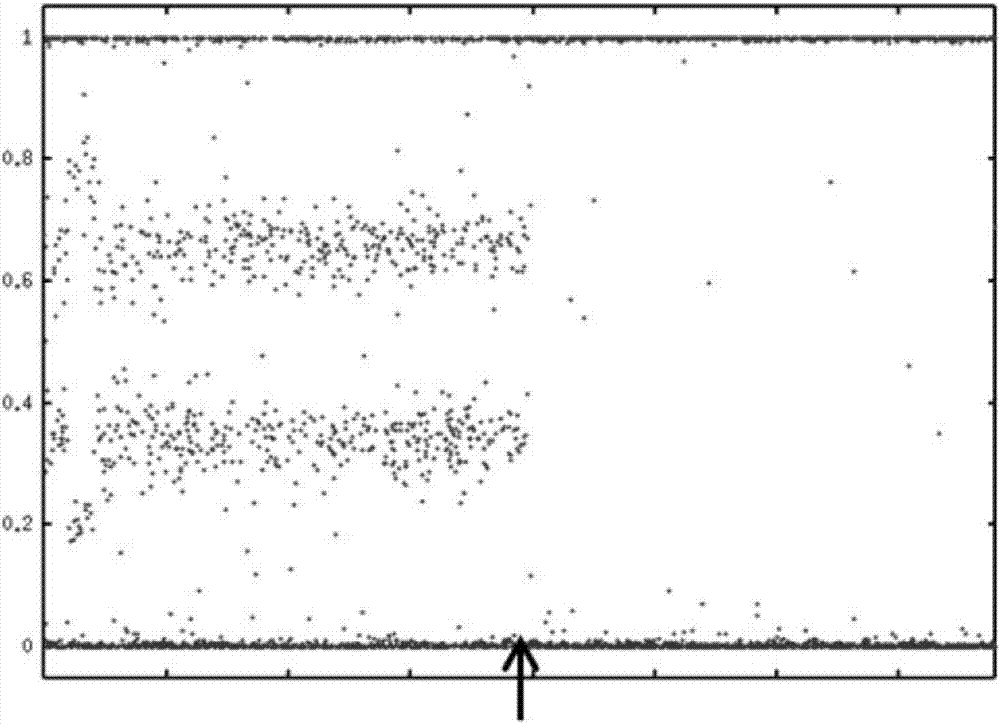
本文件相关于涉及评估样品(如:癌细胞)中是否存在杂合性丢失(loh)谱的方法和材料。例如,本文件提供了测定一个细胞(如:癌细胞)中是否包含loh谱的方法和材料。本文件还提供了鉴别有同源重组修复(hdr)缺陷的细胞(如:癌细胞)的材料和方法以及鉴别可能响应特定癌症治疗方案的癌症患者的材料和方法。在本文中,除非明确指出,hdr缺陷和hrd(同源修复缺陷)当作同义词使用。2.背景介绍癌症是一个严重的公共卫生问题,仅在2009年就有562,340的美国人口死于癌症。americancancersociety,cancerfacts&figures2009(在美国癌症协会网站可得到)。癌症治疗的主要挑战之一就是发现与患者自身癌症相关的临床上有用的特征,并且基于这些特征给出最适合患者癌症的治疗方案。尽管在个性化医疗这个领域已经有了很大的进步,但是仍然非常需要更好的分子诊断工具来特征化患者的癌症。概述一般说来,本发明的一个方面突出了评估癌细胞或其基因组dna中loh的方法。在一些实施例中,该方法包括,或大体由下列步骤组成,(a)在癌细胞或其来源的基因组dna中检测癌细胞中至少一对人类染色体上的loh区域(如:除了人类x/y性染色体对的任何一对人类染色体);和(b)测定所述loh区域的数目和大小(如:长度)。在一些实施例中,loh区域在若干代表整个基因组的染色体对中被分析(如:足够的染色体被分析,这样loh区域的数目和大小有望可以代表整个基因组的loh区域的数目和大小)。在一些实施例中,该方法进一步包括测定长于约1.5、5、12、13、14、15、16、17(优选14、14、16或更多,更优选15或者更多)百万碱基但是短于loh区域所在相应染色体整体长度的loh区域(指示loh区域)的总数。二者择一地或另外地,这些指示loh区域的总的叠加长度被测定。在一些具体的实施例中,如果指示loh区域的总数或指示loh区域的总的叠加长度等于或大于预定的参考数值,那么所述癌细胞或其基因组dna或具有所述癌细胞或基因组dna的患者被鉴定具有hdr缺陷型loh谱。还提供了一种评估癌细胞或其基因组dna中loh的另一方法,其包括,或大体由下列步骤组成,(a)在癌细胞或来源于其的基因组dna中检测癌细胞的至少一对人类染色体上的loh区域,其中所述“至少一对人类染色体”不是人类x/y性染色体对;并且(b)在所述人类染色体中测定长于第一长度的loh区域的总数和/或总的叠加长度,其中第一长度为约1.5或更多的(或5、10、13、14、15、16或更多,优选地15或更多)百万碱基,而所述loh区域短于包含该loh区域的相应的整条染色体长度。在一些实施例中,如果那个总数或叠加长度等于或大于预设的参考数,那么所述癌细胞或基因组dna或具有所述癌细胞或基因组dna的患者被鉴定具有hdr缺陷型loh标记。另一方面,本发明提供了预测癌细胞中brca1和brca2基因状态的方法。该方法包括,或大体由下列步骤组成,在癌细胞中测定癌细胞的至少一对人类染色体上的loh区域,此loh区域的长度长于第一长度的loh区域的总数和/或总的叠加长度,所述loh区域短于包含该loh区域的相应的整条染色体长度,其中所述“至少一对人类染色体”不是人类x/y性染色体对,其中所述第一长度为约1.5或更多的(或5、10或更多,优选约15或更多)百万碱基;并且大于参考数的总数或叠加长度与brca1或brca2基因缺陷的可能性的增加相关联。另一方面,本发明提供了预测癌细胞中hdr状态的方法。该方法包括,或大体由下列步骤组成,在癌细胞中测定癌细胞的至少一对人类染色体上长于第一长度的loh区域的总数和/或总的叠加长度,所述loh区域短于包含该loh区域的相应的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约为1.5或更多的(或5、10或更多,优选约15或更多)百万碱基;并且使大于参考数的总数或叠加长度与hdr缺陷的增加的可能性相关联。另一方面,本发明提供了预测癌症患者响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗和/或parp抑制剂的癌症治疗方案的方法。该方法包括,或大体由下列步骤组成,在取自癌症患者的癌细胞中测定癌症患者的癌细胞中至少一对人类染色体上的长于第一长度的loh区域的总数和/或总的叠加长度,其中第一长度为约1.5或更多的(或5、10或更多,优选约15或更多)百万碱基,而所述loh区域短于包含该loh区域的相应的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对;并且将大于参考数的总数或叠加长度与患者响应癌症治疗方案的的增加的可能性相关联。在一些实施例中,患者是未经治疗的患者。另一方面,本发明涉及预测癌症患者响应治疗方案的方法。该方法包括,或大体由下列步骤组成,在取自癌症患者的癌细胞中测定癌症患者的癌细胞中的至少一对人类染色体上长于第一长度的loh区域的总数和/或总的叠加长度,其中第一长度为约1.5或更多的(或5、10或更多,优选约15或更多)百万碱基,而所述loh区域短于包含该loh区域的相应的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对;并且将大于参考数的总数或叠加长度与患者将不响应包括紫杉醇或多烯紫杉醇的癌症治疗方案的升高的可能性相关联。另一方面,本发明有关于治疗癌症的方法。该方法包括,或大体由下列步骤组成,(a)在源自癌症患者的癌细胞或从其获得的基因组dna中测定癌细胞的至少一对人类染色体上长于第一长度的loh区域的总数和/或总的叠加长度,其中第一长度为约1.5或更多的(或5、10或更多,优选约15或更多)百万碱基,而所述loh区域短于包含该loh区域的相应的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对;并且(b)如果该loh区域的总数或叠加长度大于参考数,那么向癌症患者提供包含dna损伤剂、蒽环类药物、拓扑异构酶i抑制剂和parp抑制剂的癌症治疗方案。在一些实施例中,该患者为未经治疗的患者。在前面六段所描述的任何一个或多个方法的一些实施例中,下列中的任何一个或多个可以视情况而定被应用。loh区域可在至少二、五、十、或十二对人类染色体中被测定。癌细胞可以是卵巢、乳腺或食管癌细胞。第一长度可以是约6、12或约15或更多百万碱基(即600万、1千2百万、1千5百万或更多碱基)。该参考数可以是6、7、8、9、10、11、12、13、14、15、16、17、18或20或更大。所述至少一对人类染色体队可以排除17号人类染色体。该dna损伤剂可以是顺铂、卡铂、奥沙利铂或吡铂,该蒽环类药物可以是表阿霉素或多柔比星,该拓扑异构酶i抑制剂可以是喜树碱、拓扑替康、或伊立替康,或该parp抑制剂可以是iniparib、olaparib,或velapirib.另一方面,本发明突出了利用一种或多种选自dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂的化合物用于制备治疗确定为带有总数为5、8、9、10、12、15、17、20或更多的指示loh区域的癌细胞的患者的癌症的药物制剂。该指示loh区域可以在至少2、5、10或21对人类染色体中被测定。该癌细胞可以是卵巢癌、乳腺癌或食管癌细胞。该指示loh区域的长度为约6、12、或15或更多的百万碱基(即600万、1千2百万、或1千5百万或更多碱基。该指示loh区域可以存在于除17号人类染色体以外的染色体上。该dna损伤剂可以是铂类化疗药物,该蒽醌类可以是表阿霉素或阿霉素,该拓扑异构酶i抑制剂可以是喜树碱、拓扑替康或伊立替康,或该parp抑制剂可以是iniparib、olaparib或velapirib。在一些实施例中,该患者是未经治疗的患者。另一方面,本发明突出了利用能够与人类基因组dna的多个多态性区域杂交的多个寡核苷酸来制备用于测定从癌症患者那里得到的人类癌细胞的至少一对染色体上的指示loh区域的总数或叠加长度的诊断试剂盒,并且此诊断试剂盒还可以用于测定(a)癌细胞中brca1或brca2基因缺陷的可能性的增加,(b)癌细胞中hdr缺陷的可能性的增加,或(c)癌症患者将会响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、或parp抑制剂的治疗方案的可能性的增加。该指示loh区域可以在至少2、5、10或21对人类染色体中被测定。该癌细胞可以是卵巢癌、乳腺癌或食管癌细胞。该指示loh区域可具有约6、12或15或更多百万碱基的长度。该指示loh区域可以存在于除17号人类染色体以外的染色体上。另一方面,本发明突出了一种测定癌症患者癌细胞的loh状态的系统。该系统包括,或大体由下列组成,(a)被配置成可以产生关于癌细胞至少一对人类染色体的基因组dna的多个信号的样品分析仪,以及(b)已编程为根据所述多个信号可以计算所述至少一对人类染色体上的指示loh区域数目的计算机子系统。计算机子系统经程序控制可将指示loh区域的数目或叠加长度与参考值作比较来确定(a)癌细胞中brca1和/或brca2基因缺陷的可能性,(b)癌细胞中hdr缺陷的可能性,或(c)癌症患者将会响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、或parp抑制剂的治疗方案的可能性。该系统可包括一个被配置用来显示(a)、(b)、或(c)可能性的输出模块。该系统可包括一个被配置用来显示使用癌症治疗方案建议的输出模块。该指示loh区域可以在至少2、5、10或21对人类染色体中被测定。该癌细胞可以是卵巢癌、乳腺癌或食管癌细胞。该指示loh区域可具有约6、12、或15或更多百万碱基的长度。该指示loh区域可以存在于除17号人类染色体以外的染色体上。该dna损伤剂可以是铂-基化疗药物,该蒽醌类可以是表阿霉素或阿霉素,该拓扑异构酶i抑制剂可以是喜树碱、拓扑替康或伊立替康,或该parp抑制剂可以是iniparib、olaparib或velapirib。另一方面,本发明提供了一种体现在计算机可读介质中的计算机程序产品,当在计算机上执行时,提供了沿着除了人类x和y性染色体的一条或多条染色体检测任何loh区域的存在或缺乏的指令,并且该loh区域具有约1.5或更多(5、10或更多,优选15或更多)百万碱基但是短于包含该loh区域的整条染色体长度的长度;并且测定在一对或多对染色体中的符合上述条件的loh区域总数或叠加长度。该电脑程序产品可以包括其它的指令。该指示loh区域可以在至少2、5、10或21对人类染色体上被测定。该癌细胞可以是卵巢癌、乳腺癌或食管癌细胞。该指示loh区域可具有约6、12、或15或更多的百万碱基的长度。该指示loh区域可以存在于除17号人类染色体以外的染色体上。该dna损伤剂可以是基于铂的化疗药物,该蒽醌类可以是表阿霉素或阿霉素,该拓扑异构酶i抑制剂可以是喜树碱、拓扑替康或伊立替康,或该parp抑制剂可以是iniparib、olaparib或velapirib。另一方面,本发明提供了一种诊断试剂盒。该试剂盒包括,或大体由下列组成,能够与人类基因组dna的多个多态性区域杂交的至少500个寡核苷酸;以及本文所提供的计算机程序产品。该计算机程序产品可体现在计算机可读介质中,当在计算机上执行时,提供了在除人类x和y性染色体以外的一条或多条人类染色体中检测任何loh区域的存在或缺乏的指令,并且该loh区域长度为约1.5或更多(5、10或更多,优选地15或更多)百万碱基但是短于包含所述loh区域的整条染色体长度;以及测定该loh区域在一对或多对染色体对中的总数或叠加长度的指令。另一方面,本文件突出了评估患者癌细胞中是否存在loh谱的方法。该方法包括,或大体由下列步骤组成,(a)在癌症患者的癌细胞的至少一对人类染色体上检测多于参考数的loh区域的存在,所述loh区域长于第一长度并且短于包含所述相应loh区域的整条染色体长度,其中所述人类染色体不是人类x/y性染色体对,其中该第一长度为约1.5或更多的百万碱基,并且(b)鉴别患者的癌细胞是否具有所述loh谱。另一方面,本文件突出了一种评估患者癌细胞是否存在hdr缺陷状态的方法。该方法包括,或大体由下列步骤组成,(a)在癌症患者的癌细胞的至少一对人类染色体上检测多于参考数的下述loh区域的存在,其中任一所述loh区域长于第一长度并且短于包含所述相应loh区域的整条染色体长度的,其中所述至少一对人类染色体不是人类x/y性染色体对,其中该第一长度约为1.5或更多的百万碱基,并且(b)鉴定患者为带有hdr缺陷状态的癌细胞。另一方面,本文件突出了一种评估患者癌细胞中是否存在hdr路径中基因突变的方法。该方法包括,或大体由下列步骤组成,(a)在癌症患者的癌细胞的至少一对人类染色体上检测出有多于参考数的loh区域,任一所述loh区域的长度应当大于第一长度并且短于包含该loh区域的整条染色体的长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中该第一长度为约1.5或更多的百万碱基,并且(b)鉴定患者为带有hdr路径中基因突变的癌细胞。另一方面,本文件突出了测定是否患者有可能响应包括放射治疗、dna损伤剂、蒽环类、拓扑异构酶i抑制剂或parp抑制剂的癌症治疗方案的方法。该方法包括,或大体由下列步骤组成,(a)在癌症患者的癌细胞的至少一对人类染色体上检测多于参考数的下述loh区域的存在,其中loh区域应当长于第一长度并且短于包含相应loh区域的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中该第一长度为约1.5或更多的百万碱基,并且(b)鉴别可能响应癌症治疗方案的患者。另一方面,本文件突出了评估患者的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有loh谱的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的有大于参考数的长于第一长度但是短于包含相应loh区域的整条染色体长度的loh区域的存在表明癌细胞具有loh谱,其中所述至少一对人类染色体不是人类x/y性染色体对,其中该第一长度约为1百5万或更多的碱基,并且(b)诊断含有具有loh谱的癌细胞的患者。另一方面,本文件突出了评估患者的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有hdr缺陷状态的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的有多于参考数的长于第一长度但是短于包含相应loh区域的整条染色体长度的loh区域的存在表明癌细胞具有hdr缺陷状态,其中所述的至少一对人类染色体不是人类x/y性染色体对,其中该第一长度约为1百50万或更多的碱基,并且(b)诊断含有具有hdr缺陷状态的癌细胞的患者。另一方面,本文件突出了评估患者的方法。该方法包括,或大体由下列步骤组成,(a)测定带有hdr路径基因的基因突变的癌细胞的患者,其中癌症患者的癌细胞的至少一对的人类染色体总起来有多于参考数的长于第一长度但是短于包含相应loh区域的整条染色体长度的loh区域的存在表明癌细胞具有基因突变,其中所述的“至少一对人类染色体”不是人类x/y性染色体对,其中该第一长度约1.5或更多的百万碱基,并且(b)诊断含有具有基因突变的癌细胞的患者。另一方面,本文件突出了评估患者是否响应包含放射治疗或选自包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂药物的癌症治疗方案的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有loh标记的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度的loh区域,而所述loh区域短于包含该loh区域的相应的整条染色体长度,其中第一长度约1.5或更多的百万碱基,其中所述至少一对人类染色体不是人类x/y性染色体对,多于参考数的这种loh区域的存在表明癌细胞具有loh标记;并且(b)至少部分基于loh标记的存在,诊断有可能响应癌症治疗方案的患者。另一方面,本文件突出了一种执行患者癌细胞诊断分析的方法。该方法包括,或大体由下列步骤组成,(a)检测癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)鉴定包含具有loh标记的癌细胞的患者。另一方面,本文件突出了一种执行患者癌细胞诊断分析的方法。该方法包括,或大体由下列步骤组成,(a)检测癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)鉴定包含具有hdr缺陷状态的癌细胞的患者。另一方面,本文件突出了一种执行患者癌细胞诊断分析的方法。该方法包括,或大体由下列步骤组成,(a)检测癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)鉴定包含具有来自hdr途径基因的基因突变的的癌细胞的患者。另一方面,本文件突出了一种执行患者癌细胞诊断分析来确定是否该癌症患者有可能响应包含给予放射治疗或选自包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂组合的药物的癌症治疗方案的方法。该方法包括,或大体由下列步骤组成,(a)检测癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)鉴定有可能响应癌症治疗方案的患者。另一方面,本文件突出了一种诊断包含具有loh谱的癌细胞的患者的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有loh谱的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在表明癌细胞具有loh标记,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)诊断含有具有loh标记的癌细胞的患者。另一方面,本文件突出了一种诊断包含具有hdr缺陷状态的癌细胞的患者的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有hdr缺陷状态的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在表明癌细胞具有hdr缺陷状态,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)诊断含有具有hdr缺陷状态的癌细胞的患者。另一方面,本文件突出了一种诊断包含具有hdr途径基因的基因突变的癌细胞的患者的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有基因突变的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在表明癌细胞具有基因突变,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)诊断含有具有基因突变的癌细胞的患者。另一方面,本文件突出了一种诊断患者是否作为包含放射治疗或选自包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂药物的癌症治疗方案的候选人的方法。该方法包括,或大体由下列步骤组成,(a)测定包括具有loh标记的癌细胞的患者,其中癌症患者的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的存在表明癌细胞具有loh标记,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多的百万碱基,并且(b)至少部分基于loh标记的存在,诊断有可能响应癌症治疗方案的患者。如果没有另行规定,文中所用的所有的技术和科学术语与本专利相关领域的普通技术人员的通常理解有着相同的含义。尽管与这里提到的那些类似或相同的方法或材料可用于实践本发明,合适的材料和方法如下所述。在这里提到的所有的出版物、专利申请、专利、以及其他的参考文献通过索引的方式全部并入。在冲突的情况下,以本说明书包括定义为准。此外,这些材料、方法和实例只是说明性的,而不是意在限制本发明。本发明的一个或多个实施例的细节在说明书和以下的附图中被陈述。这些材料、方法和实例只是说明性的,而不是意在限制本发明。本发明的其它特征、目的以及优点在说明书、附图和权利要求书中是显而易见的。附图说明图1是利用snp阵列测定的来自乳腺癌患者的乳腺癌细胞的沿着1号染色体上的等位基因剂量的图。箭头表示杂合性区域和loh区域之间的转换。图2是利用高通量测序法测定的来自如图1所示的相同乳腺癌患者的乳腺癌细胞的沿着1号染色体的等位基因的剂量图。箭头表示杂合性区域和loh区域之间的转换。图3是评估细胞(如,癌细胞)基因组loh谱的示例流程的流程图。图4是可被用于实施本文中所描述的技术的计算机设备以及移动计算机设备的一个示例的图解。图5是在来自62例人类患者的卵巢癌细胞中检测到的loh区域的长度分布图。调整后的长度是指被loh区域覆盖了的染色体臂的比例。图6是具有完整的或缺陷的brca1和brca2基因的卵巢癌细胞样品的训练集合中长于15mb但短于整条染色体的loh区域的数量图。圆圈的大小与具有这样数目的loh区域的样品数量成比例。图7是图6是具有完整的或缺陷的brca1和brca2基因的卵巢癌细胞样品的训练集合和验证集合中长于15mb但短于整条染色体的loh区域的数量图。圆圈的大小与具有这样数目的loh区域的样品数量成比例。图8是具有体细胞brca突变、生殖系brca突变、低brca1表达或完整brca(brca正常)的卵巢癌细胞样品中长于15mb但短于整条染色体的loh区域的数量图。圆圈的大小与具有这样数量的loh区域的样品数量成比例。图9是一张表格展示brca缺陷的、hdr缺陷的/brca完整的、以及hdr完整的卵巢癌细胞样品的百分比。图10是所示癌症的肿瘤细胞系中长于15mb但短于整条染色体的loh区域的数量图。圆圈的大小与具有这样数量的loh区域的样品数量成比例。图11是肺癌样品中长于15mb但短于整条染色体的loh区域的数量图。图12是所示癌症或癌细胞系具有hdr缺陷的百分比图。图13包括当暴露于具有所示数量的长于15mb但短于整条染色体的loh区域的29例乳腺癌细胞系后喜树碱(camptothecin)的ic50值(log10(ic50)图以及铂化合物(奥沙利铂、顺铂和卡铂)、或蒽环类(阿霉素和表阿霉素)的平均log10(ic50)值图,或者当暴露于具有所示数量的长于15mb短于整条染色体的loh区域的卵巢癌细胞系后紫杉醇的ic50值(log10(ic50))图。虚线将域值数设置在9。图14是图13的标记版本,它将暴露于具有所示数量的长于15mb短于整条染色体的loh区域的29例乳腺癌细胞系后的铂化合物(奥沙利铂、顺铂和卡铂)的平均log10(ic50)值列图。图15是鉴定loh位点和区域的示例计算过程的流程图。图16展示了loh区域长度与以染色体臂长度校正的这些loh区域的长度的比值。在这个图上的最大的校正值等于2,与loh对整条染色体比值相对应。图17展示了肿瘤样品的hrd分值。蓝圈:brca1或brca2缺陷样品。红圈:brca1和brca2完整样品。蓝圈和红圈的合并区域是相同的。每个单独的圈的面积与具有相应数目的loh区域的样品数呈比例。图17a.第一组的hrd分值(152例样品中的46例是brca1或brca2缺陷)。图17b.第二组的hrd分值(53例样品中的19例是brca1或brca2缺陷)。图17c.第三组的hrd分值(435例样品中的146例是brca1或brca2缺陷)。图17d.来自所有三组的总合的数据的hrd分值。a行:224例样品有brca1、或brca2、或rad51c基因缺陷;b:84例brca1突变体;c:43例brca2突变体;d:82例样品有brca1低表达或甲基化;e:13例样品具有rad51c甲基化。红圈:416例样品具有brca1、brca2和rad51c完整基因。图18a.癌细胞系中hrd分值的比较。红圈:具有完整brca1或brca2的细胞系。a:30例完整的非卵巢细胞系;b:22例完整的卵巢细胞系。绿圈:6例具有brca1或brca2杂合突变的携带者。紫圈:2例具有brca1或brca2隔代遗传的杂合突变的携带者。蓝圈:7例具有brca1或brca2杂合突变或甲基化brca1的携带者。绿、红、蓝和紫圈的合并区域是相同的。每个单独的圈的面积与具有相应数目的loh区域的样品数呈比例。图18b.手术后生存期的hrd值的kaplan-meier图谱在中线处拆分。这些数据是用具有拷贝数数据和生存资料的tgca数据集的507例样品生成的。具有高和低hrd分值的样品的中位生存期分别是1499(95%ci=(1355-1769))和1163(95%ci=(1081-1354))天。图19显示了第一研究组中loh分值与用不同loh区域长度的截止阈值计算的hr缺陷之间的相关性。相应的log10(p-值)如y-轴所示。研究了loh区域长度的截止阈值和hr缺陷与loh分值相关的显著性之间的关系。该图显示loh长度截止阈值可以是11-21mb。以15mb作为截止阈值,约在间隔的中间,可以用在一些优选的实施例中,因为它被认为对存在于该数据中的统计噪音更敏感。图20显示了三组brca1和brca2缺陷样品中loh分值的比较,来自所有三个研究组群的叠加数据。a行:49例brca1生殖系突变携带者;a:25例brca1体细胞突变携带者;c:82例brca1甲基化或低表达携带者;d:27例brca1生殖系突变携带者;e:9例brca2体细胞突变携带者。图21显示了brca1、brca2和rad51c缺陷样品的loh分值的比较关系。蓝圈对应于brca1缺陷样品,红圈对应于brca2缺陷样品,绿圈对应于rad51c缺陷样品。红、蓝和绿圈下的合并区域是相同的。每个单独的圈的面积与具有相应数目的loh区域的样品数量呈比例。发明详述本文件提供了用于评估样品(如:癌细胞)中是否存在loh谱的方法和材料。例如,本文件提供了测定一个细胞(如:癌细胞)中是否包含loh谱(如,hdr缺陷loh谱)的方法和材料。一般说来,存在于每条染色体(男性的每条常染色体)相同位点的序列的比对关系可揭露是否这个在细胞基因组中的特定的位点是纯合的或杂合的。在人类基因组内的多态性基因位点在一个个体内一般都是杂合的,因为个体通常接收来自生父的一份拷贝和来自生母的一份拷贝。在某些情况下,在一个个体内的一个多态性基因位点或一连串多态性基因位点是纯合的是因为从亲生父母继承相同的拷贝。杂合性丢失(loh)可能起因于很多机制。例如,在某些情况下,一条染色体的一个区域可以在体细胞中缺失。仍然存在于另一条染色体(男性的其它非性染色体)上的该区域是一个loh区域,因为只有该区域的一个拷贝(而不是两个)存在于受影响的细胞的基因组内。这个loh区域可是是任何长度(如,从少于约1.5mb的长度到等于染色体整体长度)。这种类型的loh事件导致了拷贝数的减少。在其它情况下,一条染色体(男性的非性染色体)上的一个区域在体细胞中被来自另一染色体的该区域的一个拷贝所替代,因此消除了可能存在于被替代区域的任何的杂合性。在这种情况下,仍然存在于每条染色体上的该区域是一个loh区域并且可以简称为一个拷贝中性loh区域。拷贝中性loh区域可以是任何长度(如,从少于约1.5mb的长度到等于染色体整体长度)。如本文中所述,一个细胞样品(如,癌细胞样品)可以被鉴定为含有“阳性loh标记状态”(或者称为“hdr缺陷型loh标记”),如果该细胞的基因组被评估含有5个或更多(如,6或更多、7或更多、8或更多、9或更多、10或更多、11或更多、12或更多、13或更多、14或更多、15或更多、16或更多、17或更多、18或更多、19或更多、或20或更多)loh区域,该loh区域(a)长于约1百50万碱基(如,长于2、2.5、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、35、40、45、50、75或100百万碱基(mb),优选地长于14或15或16,更优选地长于约15百万碱基),并且(b)少于包含该loh区域的整条染色体的长度。在某些情况下,一个癌细胞样品可以被认为含有阳性loh标记状态,如果该细胞的基因组被评估含有9个或更多loh区域,该loh区域(a)长于约15mb并且(b)少于包含该loh区域的整条染色体的长度。除非另有说明,术语“指示loh区域”指的是在除人类x/y性染色体对以外的一对染色体上的并且具有杂合性丢失特征的长度约1百50万或更多碱基但是短于含有该指示loh区域的整条染色体的长度的loh区域。包含loh区域的整条染色体的长度可以通过检查生殖细胞或非肿瘤体细胞中对应的染色体对的较短的染色体长度测定的。在一些实施例中,一个指示loh区域是任何约2、2.5、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、35、40、45、50、75或100个百万碱基(mb)或更多(优选长于14或15百万碱基,即大于1千4百万或1千5百万碱基)但小于包含该loh区域的整条染色体长度的loh区域。被认定为具有阳性loh标记(在这里也被称为“hdr缺陷loh标记”)的细胞(如,癌细胞)可被归类于具有hdr缺陷可能性的增加和/或具有hdr途径一个或多个基因缺陷状态可能性的增加。例如,被认定为具有阳性loh标记的癌细胞可被归类于具有hdr缺陷状态可能性的增加。在某些情况下,被认定为具有阳性loh标记的癌细胞可被归类于具有hdr途径一个或多个基因缺陷状态可能性的增加。在本文中用到的,基因的缺陷状态意思是基因的序列、结构、表达和/或活性或它的产物与正常相比是有缺陷的。例如其中包括但不限于,低或无mrna或蛋白质表达、有害突变、超甲基化、弱活性(如,酶活性、结合其它生物分子的活性)等。在本文中用到的,途径(如,hdr途径)的缺陷状态意思是这个途径中的至少一个基因(如,brca1)是有缺陷的。特别有害的突变的例子包括移码突变、终止子突变、以及导致rna剪接改变的突变。hdr途径的基因的缺陷状态可以导致癌细胞中同源定向修复的缺陷或减活。hdr途径基因的例子包括但不限于,表1所列基因。表1.部分hdr途径基因存在于hdr途径中的基因的基因突变的例子包括但不限于,表2所列基因。表2.在所选的hdr途径的基因中可能的基因突变在某些情况下,一个细胞样品(如,癌细胞样品)可被鉴定为含有数量增加的覆盖整条染色体的loh区域(如,至少7、8、9、10或更多的loh区域)。被鉴定为含有数量增加的覆盖整条染色体的loh区域的细胞(如,癌细胞)可被归类于具有hdr能力也就是完整hdr途径的可能性的增加。例如,被鉴定为具有数量增加的覆盖整条染色体的loh区域的癌细胞可被归类于更有可能含有完整的brca1和brca2基因。如本文中所述,鉴定loh位点(以及loh区域的大小和数量)可包括,首先,确定样品在多个基因组位点(如,snp位点,单个碱基的大规模测序)的基因型以及,第二,确定是否纯合位点归因于loh事件。任何合适的技术可用来确定在一个细胞基因组内感兴趣位点的基因型。例如,单核苷酸多态性(snp)阵列(如,人类基因组范围的snp阵列)、感兴趣位点的目标测序(如,snp位点以及它们周围序列的测序)以及甚至非目标测序(如,整个外显子组、转录组或基因组测序)可用来鉴定位点是纯合的或是杂合的。在某些情况下,可执行覆盖一条染色体长度的位点的纯合的或杂合特性的分析来确定纯合性或杂合性区域的长度。例如,利用snp阵列结果评估的一段沿着一条染色体的已定距离间隔(如,间隔约25kb到约100kb)的snp位置来确定沿着染色体的纯合区域的存在以及该区域的长度。来自snp阵列的结果可用来产生绘制等位基因沿染色体的剂量图。snpi的等位基因剂量di可从调整过的两个等位基因的信号强度计算出来(ai和bi):di=ai/(ai+bi)。这样一个图表的示例展示在表图1中。对本发明有用的核酸阵列的许多不同形式在本领域是已知的。这些包括下面的各个示例中所使用的阵列(如,示例3中的affymetrix500k基因芯片分析;示例4中的affymetrixoncoscantmffpeexpress2.0services(以前的mipcnservices)).一旦一个样品的多个位点基因型被确定(如,snps),常用的技术可以用来识别loh的位点和区域。确定是否纯合归因于loh的一种方法是将体细胞基因型和生殖系基因型进行比较。例如,可在生殖系(如,血液)样品和体细胞(如,肿瘤)样品中确定多个位点(如,snps)的基因型。比较每个样品的基因型(一般用计算机)来确定生殖系细胞基因组是杂合而体细胞基因组却是纯合的位点,这样的位点是loh位点,这样的位点的区域是loh区域。计算技术也可以用于确定是否纯合性归因于loh。当没有生殖系样品可用来分析和比较时,这样的技术是非常有用的。例如,那些在别处描述的算法可以用来自snp阵列(nannyaetal.,cancerres.(2005)65:6071-6079(2005))的信息来检测loh区域。通常,这些算法并没有明确考虑到良性组织对肿瘤样品的污染。cf.国际申请no.pct/us2011/026098toabkevichetal.;goranssonetal.,plosone(2009)4(6):e6057。这种污染通常高到足以使loh区域的检测受到质疑。根据本发明用于鉴定loh的改良的分析方法,甚至在有污染的情况下,包括那些如下所述计算机软件产品。下面是一个示例。如果观察到的两个等位基因的信号比,a和b,是2到1,有两种可能性,第一种可能性是在被50%正常细胞污染的样品中具有loh的癌细胞中有等位基因b缺陷。第二种可能性是在没有被正常细胞污染的样品中没有loh但是有等位基因a的复制。一种可通过本文描述的电脑程序来实施的算法可用来重建基于基因型(如,snp基因型)数据的loh区域。该算法的一个点是首先重建每个位点(如,snp)的等位基因特异性拷贝数(ascn)。ascns是父系和母系等位基因的拷贝数目。如果一个snps区域的ascns(父系或母系)中的一个是0,则确定为一个loh区域。该算法可以基于最大化似然函数,可能在概念上类似于之前描述的被设计用来重建每个位点(如,snp)总拷贝数的算法(见国际专利no.pct/us2011/026098toabkevichetal)。似然函数可将所有位点的ascn、良性组织污染程度、整个基因组的平均总拷贝数、以及样品特异性噪声水平最大化。该算法的输入数据可以包括或包含(1)每个位点两个等位基因的样品特异性标准化信号强度和(2)基于对大量已知ascn谱的样品的分析来确定的分析方法特异性的参数组(针对不同的snp阵列和基于序列的方法)。在某些情况下,核酸测序技术可被用来识别位点是纯合或杂合的。例如,来自细胞样品(如,癌细胞样品)的基因组dna可以被提取和片段化。可用来提取和片段化基因组核酸的任何合适的方法包括但不限于商品化试剂盒如qiaamptmdnaminikit(qiagentm)、magnatmpurednaisolationkit(rocheappliedsciencetm)以及genelutetmmammaliangenomicdnaminiprepkit(sigma-aldrichtm)。一旦提取和片段化后,开展目标化或非目标化测序来确定样品在位点处的基因型。例如,可开展整个基因组、整个转录组、或整个外显子组测序来确定数百万甚至数十亿个碱基对的基因型(如,碱基对可以作为“位点”进行评估)。在某些情况下,可开展已知的多态位点(如,snps及其周围序列)的靶向测序作为微阵列分析的一种替代。例如,用为此目的而设计的试剂盒(如,agilentsureselecttm、illuminatruseqcapturetm和nimblegenseqcapezchoicetm)富集那些基因组dna的包含被分析位点(如,snp位置)的片段。例如,将包含被分析位点的基因组dna与生物素化捕获rna片段杂交形成生物素化rna/基因组dna复合物。或者,dna捕获探针可被用于导致形成生物素化rna/基因组dna杂交体。链霉亲和素包被的磁珠以及磁力可被用来从那些不存在于生物素化rna/基因组dna复合物中的基因组dna片段中分离生物素化rna/基因组dna复合物。处理所得到的生物素化rna/基因组dna复合物,从磁珠上去除捕获的rna,从而留下包含被分析位点的完整的基因组dna片段。用如pcr技术扩增这些包含被分析位点的完整的基因组dna片段。用高通量测序技术或新一代测序技术如illuminahiseqtm,illuminamiseqtm或美国lifetechnologies的solidtm或iontorrenttm或roche454tm来测序扩增了的完整的基因组dna片段。来自基因组dna片段的测序结果可被用来识别位点是纯合的或是杂合的,类似于本文所述的微阵列分析。在某些情况下,可以开展执行一条染色体长度的位点的纯合或杂合特性的分析来确定纯合或杂合区域的长度。例如,一段沿着一条染色体的给定距离间隔(如,间隔约25kb到约100kb)的snp位置通过测序被评估,该测序结果用来确定沿着染色体的纯合区域的存在以及该区域的长度。得到的测序结果可用来绘制等位基因沿染色体的剂量图。snpi的等位基因计量di可从调整过捕获探针的数量的两个等位基因的计算出来(ai和bi):di=ai/(ai+bi)。这样一个图表的示例展示在图2中。根据本文所述实施确定是否纯合归因于loh(生殖系纯合作为对照)。在某些情况下,一个筛选过程可被用来选择被评估位点(如snp位点),这一筛选过程可利用一种鉴定位点是纯合或杂合的方法(如,基于snp阵列的试验及基于测序的试验)。例如,任何人类snp位置可以被选出来并用于基于snp阵列的试验或基于测序的试验中,这些试验可以用来鉴定细胞基因组中的位点为纯合的或杂合的。在某些情况下,存在于人类基因组的0.5百万、1.0百万、1.5百万、2.0百万、2.5百万或更多snp位置可被评估用来鉴定那些(a)不存在于y染色体,(b)不为线粒体snps,(c)在白种人中具有至少约5%的次要等位基因频率,(d)在除白种人以外的三个种族(如,中国人、日本人、以及约鲁巴人)中具有至少约1%的次要等位基因频率,和/或(e)在这四个种族中没有与哈迪温伯格平衡的显著差异。在某些情况下,多于100,000、150,000或200,000的满足标准(a)-(e)的人类snps可被筛选出来。在满足标准(a)-(e)的人类snps中,在白种人中具有高等位基因频率的这样一组snps(如,最多110,000snps)可被筛选出来,它以稍微均匀间隔的方式(如,约每25kb-500kb至少一个snp)覆盖在人类基因组中,并且与从四个种族中的任何一个筛选出的其它的snp没有连锁不平衡。在某些情况下,约4万、5万、6万、7万、8万、9万、10万、11万、12万、13万或更多的满足所有这些标准snps可被筛选出来并可被包括在设定的用来鉴定跨越人类染色体组的loh区域的试验中。例如,约70,000-90,000(如,约80,000)之间的snps可被筛选出来用于基于snp阵列的试验,并且约45,000-55,000(如,约54,000)snps之间的snps可被筛选出来用于基于测序的试验。如文中所描述的,可以评估细胞样品来确定样品细胞的基因组是否含有loh标记,缺少loh标记,具有数量增加的loh区域覆盖整条染色体的,或覆盖整条染色体的loh区域数量没有增加。可以评估任何合适类型的样品。例如,可以评估含有癌细胞的样品来确定样品细胞的基因组是否含有loh标记,缺少loh标记,覆盖整条染色体的loh区域数量增加,或覆盖整条染色体的loh区域没有数量增加。在这里描述的可被评估的包含癌细胞的样品的示例包括但不限于肿瘤活体样品(如,乳腺肿瘤活体样品)、包含癌细胞的福尔马林固定的、石蜡包埋的组织样品、活组织检查、细针抽出物、以及含有从肿瘤脱落的癌细胞的样品(如,血液、尿液或其它体液)。对于福尔马林固定的、石蜡包埋的组织样品来说,样品可以通过dna提取采用ffpe组织优化的基因组dna提取试剂盒制备,这些试剂盒包括但不限于前面所述的那些(如,快提fpedna提取试剂盒(epicentretm)和qiaamptmdnaffpe组织试剂盒(qiagentm)。在一些情况下,可以对组织样品实施激光剥离技术使被评估癌细胞样品中非癌细胞的数量最小化。在一些情况下,基于纯化方法的抗体可被用于富集癌细胞和/或去除非癌细胞。可用于癌细胞富集的抗体的示例包括但不限于anti-epcam、anti-trop-2、抗-c-met、抗叶酸结合蛋白、抗-n-cadherin、抗-cd318、抗-间叶细胞的干细胞抗原、抗-her2、抗-muc1、抗-egfr、抗-cytokeratins(如,细胞角蛋白7、细胞角蛋白20等),抗-caveolin-1、抗-psa、抗-ca125以及抗-surfactantprotein抗体。任何类型的癌症细胞可以使用本文描述的方法和材料进行评估。例如,可评估乳腺癌细胞、卵巢癌细胞、肝癌细胞、食管癌细胞、肺癌细胞、头颈癌细胞、前列前癌细胞、结肠癌细胞、直肠癌细胞或结直肠癌细胞以及胰腺癌细胞来确定样品细胞的基因组是否含有loh标记,缺少loh区域,覆盖整条染色体的loh区域数量增加,或覆盖整条染色体的loh区域数量未增加。在一些实施例中,癌细胞是卵巢癌、乳腺癌、肺癌或食道癌的原发性或转移性肿瘤细胞。当评估癌细胞的基因组是否存在或缺乏loh标记时,一或多(如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22或23)对染色体可被评估。在一些实施例中,用一或多(如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22或23)对染色体来评估癌细胞的基因组是否存在或缺乏loh标记。在一些实施例中,排除某些染色体会有益处。例如,在女性的情况下,被评估的一对可包括x性染色体对;然而,在男性的情况下,任何一对常染色体(如,除了x和y性染色体外的任何染色体对)可被评估。在另一个例子中,在某些实施例中,17号染色体对可从分析中被排除。已经确定某些染色体在某些癌症中携带异常高水平的loh,并且,当分析文中所述的来自具有这些癌症的患者的样品时,排除这样的染色体是有帮助的。在某些情况下,样品来自具有卵巢癌的患者,并且被排除的染色体是17号染色体。当评估癌细胞的基因组中覆盖整条染色体的loh区域数量是否增加时,10或更多(如,13、16、19或23)对染色体可被评估。在女性的情况下,被评估的可包括x性染色体对;然而,在男性的情况下,任何一对常染色体(如,除了x和y性染色体的任何染色体对)可被评估。在某些实施例中,17号染色体对可从分析中被排除。在某些实施例中,样品来自具有卵巢癌的患者,并且被排除的染色体是17号染色体。在某些实施例中,用10或更多(如,13、16、19或23)对染色体来评估癌细胞的基因组中覆盖整条染色体的loh区域的数量是否增加。因此,可分析预先给定数量的染色体来确定指示loh区域的总数,优选长度大于9、10、13、14百万碱基,更优选大于1千5百万碱基的loh区域的总数。或另外,可把所有被鉴定的指示loh区域的大小叠加总和来得到指示loh区域的总长度。为了确定阳性loh标记状态,上述指示loh区域的总数的参考数可以是5、6、7、8、9、10、11、12、13、14、15、16、18、19、20或更大,优选5、优选8,更优选9或10,最优选10。指示loh区域的总(如,叠加)长度的参考数可以是约75、90、105、120、130、135、150、175、200、225、250、275、300、325、350、375、400、425、450、475、500百万碱基或更大,优选约7千5百万碱基或更大,优选9千万或1亿零5百万碱基或更大,更优选1亿2千万或1亿3千万碱基或更大,更优选约1亿3千5百万碱基或更大,并且更优选1亿5千万碱基或更大。在一些具体的实施例中,确定长度大于约1千4百万或1千5百万碱基的loh区域的总数并且与约5、6、7、8、9、10、11、12、13、14、15、16、18、19或20的参考数做比较。或者,确定长度大于约1千4百万或1千5百万碱基的loh区域的总长度,并与约75mb、90mb、105mb、120mb、130mb、135mb、150mb、175mb、200mb、225mb、250mb、275mb、300mb、325mb、350mb、375mb、400mb、425mb、450mb、或500mb的参考数作比较。在某些实施例中,如果loh区域的数量(或叠加长度、或检验值或来自其中之一的得分)至少2-、3-、4-、5-、6-、7-、8-、9-或10-倍大于参考值,则认为患者样品中loh区域的数量(或叠加长度、或检验值或来自其中之一的得分)“大于”参考数。而在其它实施例中,如果比参考值至少大1、2、3、4、5、6、7、8、9或10个标准差,则认为是“大于”参考值。反之,在某些实施例中,如果loh区域的数量(或叠加长度、或检验值或来自其中之一的得分)没有比参考值至少大2-、3-、4-、5-、6-、7-、8-、9-或10-倍,则认为患者样品中loh区域的数量(或叠加长度、或检验值或来自其中之一的得分)“不大于”参考数,而在其它实施例中,如果没有比参考值至少大1、2、3、4、5、6、7、8、9或10个标准差,则认为是“不大于”参考值。在一些实施例中,参考数(或长度、值或得分)来源于相关的参考人群。这样的参考人群可能包括(a)具有与被检测患者相同的癌症,(b)具有相同的癌症亚型,(c)具有相似的遗传或其它临床或分子特征的癌症,(d)响应特定治疗的人,(e)不响应特定治疗的人,(f)表观健康的人(如,没有任何癌症或没有被检测患者的癌症)等。参考数(或长度、值或得分)可能是(a)代表在作为整体的参考人群中发现的数值(或长度、值或得分),(b)在作为整体的参考人群或特定的亚群中发现的数值(或长度、值或得分)的平均值(平均值、中值、等),(c)代表在归类于(i)它们的代表数(或长度、值或得分)或(ii)他们被发现有的临床特征(如,响应的强度、预后(包括时间到癌症特异性死亡)等)的参考人群的百分位点、四分位点、五分位点等中发现的值(或长度、值或得分)(平均如平均值、中值)。如本文中所描述,如果患者带有被检测为有阳性loh标记状态的癌细胞,这样的患者可以至少部分基于阳性loh标记状态鉴定为很可能响应特定癌症治疗方案。例如,如患者带有被检测为有阳性loh标记状态的基因组的癌细胞,这样的患者可以至少部分基于阳性loh标记状态鉴定为很可能响应包括dna损伤剂、合成致死剂(如,parp抑制剂)、放射治疗或它们的组合的特定癌症治疗方案。该患者优选地为未经治疗的患者。dna损伤剂的示例包括但不限于,基于铂的化疗药物(如,顺铂、卡铂、奥沙利铂、及吡铂),蒽醌类(如,表阿霉素和阿霉素),拓扑异构酶i抑制剂(如,喜树碱、拓扑替康、伊立替康),dna交联剂如丝裂霉素c,以及三氮烯化合物(如,达卡巴嗪和替莫唑胺)。合成致死治疗方案通常涉及给予患者对特定肿瘤细胞存活非常重要的生物路径的至少一种关键部件的抑制剂。例如,当一个肿瘤细胞具有缺陷的同源修复路径(如,根据本发明确定的)时,多聚adp核糖聚合酶抑制剂(或铂类药物、双链断裂修复抑制剂、等)可以对这些肿瘤特别有效,因为对存活至关重要的两种途径(一种生物学的,如,通过brca1突变,及另一种合成性的,如,给予一种路径药物)被阻断了。如在o’brienetal.,convertingcancermutationsintotherapeuticopportunities,embomol.med.(2009)1:297-299中描述了癌症治疗的合成致死方法。合成致死剂的例子包括但不限于,parp抑制剂或双链断裂修复抑制剂用于同源修复缺陷肿瘤细胞、parp抑制剂用于pten-缺失肿瘤细胞、甲氨蝶呤用于msh2缺失肿瘤细胞等。parp抑制剂的例子包括但不限于olaparib、iniparib以及veliparib。双链断裂修复抑制剂的例子包括但不限于ku55933(atm抑制剂)和nu7441(dna-pkcs抑制剂)。除了阳性loh标记状态,可以辅助判断患者是否能响应特定的癌症治疗方案的信息包括但不限于,以前的治疗结果、生殖细胞或体细胞dna突变、基因或蛋白质表达谱(如,er/pr/her2状态、psa水平)、肿瘤组织学(如,腺癌、鳞状细胞癌、浆液性乳头状癌、粘液癌、浸润性导管癌、非浸润性导管原位癌等)、疾病分期、肿瘤或癌症等级(如,高、中间、或低分化(如,gleason,modifiedbloomrichardson)等)、以往疗程的数量等。一旦被归类为可能响应特定的癌症治疗方案(如,包括应用dna损伤剂、parp抑制剂、放射治疗或其组合的癌症治疗方案),癌症患者可用如此的癌症治疗方案治疗。在一些实施例中,该患者是未经治疗的患者。治疗所讨论癌症的任何合适的方法可被用来治疗被鉴定为含有具有阳性loh标记状态的癌细胞的癌症患者。例如,铂类化疗药物或铂类化疗药物的组合物可被用来治疗癌症,正如其它地方描述的(如,美国专利号3,892,790、3,904,663、7,759,510、7,759,488和7,754,684)。在某些情况下,蒽环类或蒽环类的组合物可被用来治疗癌症,正如其它地方描述的(如,美国专利号3,590,028、4,138,480、4,950,738、6,087,340、7,868,040和7,485,707)。在某些情况下,拓扑异构酶i抑制剂或拓扑异构酶i抑制剂的组合物可被用来治疗癌症,正如其它地方描述的(如,美国专利号5,633,016和6,403,563)。在某些情况下,parp抑制剂或parp抑制剂的组合物可被用来治疗癌症,正如其它地方描述的(如,美国专利号5,177,075、7,915,280和7,351,701)。在某些情况下,放射治疗可用来治疗癌症,正如其它地方描述的(如,美国专利号5,295,944)。在某些情况下,由不同制剂(如,由铂-基化疗药物、蒽环类、拓扑异构酶i抑制剂、和/或parp抑制剂中的任何一个或一些组成的组合物)以及有或没有放射治疗治疗组成的组合物可被用于治疗癌症。在某些情况下,组合的治疗可能包括任何上述的化合物或治疗(如,dna损伤剂、parp抑制剂、放射治疗或它们的组合)以及其它的化合物或治疗一起,如紫杉类化合物(如,多西他赛、紫杉醇、紫杉醇(白蛋白结合型))、生长因子或生长因子受体抑制剂(如,厄洛替尼、吉非替尼、拉帕替尼、舒尼替尼、贝伐单抗、西妥昔单抗、曲妥珠单抗、帕尼单抗),和/或抗代谢物(如,5-氟尿嘧啶、氨甲喋呤)。在某些情况下,如果患者癌细胞基因组被检测为缺乏loh标记,至少部分基于阴性loh标记,患者可被鉴定为不太可能响应包含dna损伤剂、parp抑制剂、放射治疗或其组合的治疗方案。相应的,这样的患者可被鉴定为可能响应包括一种或多种与hdr不相关的癌症治疗药物(如,紫杉类化合物(如,多西他赛、紫杉醇、紫杉醇(白蛋白结合型))、生长因子或生长因子受体抑制剂(如,厄洛替尼、吉非替尼、拉帕替尼、舒尼替尼、贝伐单抗、西妥昔单抗、曲妥珠单抗、帕尼单抗),和/或抗代谢物(如,5-氟尿嘧啶、氨甲喋呤))的癌症治疗方案。在一些实施例中,患者为未经治疗的患者。一旦被归类为可能响应特定的癌症治疗方案(如,包括与hdr不相关的癌症治疗制剂的癌症治疗方案),癌症患者可用如此的癌症治疗方案治疗。治疗癌症的任何合适的方法可被用来治疗被鉴定为含有具有阴性loh标记状态的癌细胞的癌症患者。除了阴性loh标记状态,其它可参考的信息包括但不限于,以前的治疗结果、生殖细胞或体细胞dna突变、基因或蛋白质表达谱(如,er/pr/her2状态、psa水平)、肿瘤组织学(如,腺癌、鳞状细胞癌、浆液性乳头状癌、粘液癌、浸润性导管癌、非浸润性导管原位癌等)、疾病分期、肿瘤或癌症等级(如,高、中间、或低分化(如,gleason,modifiedbloomrichardson)等)、以往疗程的数量等。治疗一段时期后(如,一到六个月之间),可评估这些患者来确定治疗方案是否有效。如果检测到有益效果,患者可用相同或类似的癌症治疗方案继续治疗。如果检测到很小的或没有检测到有益效果,需要对癌症治疗方案做出调整。例如,可增加剂量、给药的频率或治疗的持续时间。在某些情况下,其它的抗癌制剂可加入到治疗方案中,或用一种或多种不同的抗癌药代替特定的抗癌药物。被治疗的患者可以继续适当的监控,并且癌症治疗方案可做些合适的改变。除了预测可能的治疗响应或选择理想的治疗方案,loh标记可被用来确定患者的预后。如下面示例3(尤其是图18b)所示,相比较肿瘤没有这种loh标记的患者来说,肿瘤具有loh标记的患者表现出显著更好的存活。因此,一方面,本文档突出了测定患者预后的方法,该方法至少一部分基于在来自患者的样品中检测loh标记的存在或缺乏。该方法包括,或大体由下列步骤组成,(a)测定患者是否包括具有文中描述的loh标记(如,其中,癌症患者的癌细胞的至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的多于参考数的loh区域的存在表明癌细胞具有loh标记,其中所述至少一对人类染色体不是人类x/y性染色体对,其中该第一长度约1.5或更多的百万碱基)的癌细胞,并且(b)(1)至少部分基于loh标记的存在,确定具有相对好的预后的患者,或(b)(2)至少部分基于loh标记的缺乏,确定具有相对不良预后的患者。预后可能包括患者存活的可能性(如,无恶化存活期,总存活率),其中相对好的预后可能包括,相比一些参考人群(如,具有这种患者癌症类型/亚型的平均患者,不具有loh标记的平均患者,等),具有增加的存活可能性。相反地,依据存活率的相对不良预后可能包括,相比一些参考人群(如,具有这种患者癌症类型/亚型的平均患者,具有loh标记的平均患者,等),具有降低的存活可能性。如本文中所描述,本文件提供了预测具有包含loh标记的基因组的细胞(如,癌细胞)的患者的方法。在一些实施例中,患者是未经治疗的患者。例如,一个或多个临床医生或医疗专业人员可确定是否一个患者包括具有包含loh标记的基因组的癌细胞。在某些情况下,一个或多个临床医生或医疗专业人员可通过从患者获得癌细胞样品以及评估癌细胞样品的癌细胞基因组来测定文中描述的loh标记的存在或缺乏来确定是否一个患者包括具有包含loh标记的基因组的癌细胞。在某些情况下,一个或多个临床医生或医疗专业人员可从患者那里获得癌细胞样品并且给样品提供一个具有评估癌细胞样品的癌细胞基因组来提供关于文中描述的loh标记存在或缺乏的指示的能力的测试实验室。在一些实施例中,患者是未经治疗的患者。在这样的情况下,一个或多个临床医生或医疗专业人员可通过直接或间接从测试实验室接收关于loh标记存在或缺乏的信息来确定是否一个患者包括具有包含loh特性的基因组的癌细胞。例如,一个测试实验室,在评估了癌细胞基因组中如前描述的loh标记的存在或不存在之后,可以给一个或多个临床医生或医疗专业人员提供或使他们有权使用关于被评估的特定患者的loh标记存在或缺乏的信息指示的书面、电子或口头报告或病历。这样的书面、电子或口头报告或病历可以允许一个或多个临床医生或医疗专业人员确定一个特定的被评估患者是否具有包含loh标记的基因组的癌细胞。一个临床医生或医疗专业人员或一组临床医生或医疗专业人员确定了特定的被评估的患者包括具有包含loh标记的基因组的癌细胞,该临床医生或医疗专业人员(或一组)可以鉴定具有癌细胞的患者,该癌细胞的基因组包括loh标记的存在。在一些实施例中,患者是未经治疗的患者。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将已确定具有包含loh标记存在的基因组的癌细胞的患者诊断为具有可能hdr缺陷的癌细胞。这样的诊断可以唯一地基于包括具有包含loh标记的基因组癌细胞的特定的被评估患者的检测或至少部分的基于包括具有包含loh标记的基因组癌细胞的特定的被评估患者的检测。例如,基于一个或多个肿瘤抑制基因(如,brca1/2、rad51c)中的阳性loh标记状态和缺陷状态、癌症的家族史、或行为危险因素(如,吸烟)的存在的联合,已被确定具有包括loh标记存在的基因组的癌细胞的患者可被诊断为可能是hdr缺陷。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将已确定具有包含loh标记存在的基因组的癌细胞的患者诊断为具有可能包含hdr途径中一个或多个基因基因突变的癌细胞。在一些实施例中,患者是未经治疗的患者。这样的诊断可以仅仅基于被评估患者具有包含loh标记的基因组的癌细胞或至少部分的基于被评估患者具有包含loh标记的基因组癌细胞。例如,基于阳性loh标记状态和癌症的家族史、或行为危险因素(如,吸烟)的存在的联合,已被确定具有包括loh标记存在的基因组的癌细胞的患者可被诊断为可能具有包含hdr途径中一个或多个基因基因突变的癌细胞。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将已确定具有包含loh标记存在的基因组的癌细胞的患者诊断为具有可能响应特定癌症治疗方案的癌细胞。在一些实施例中,患者是未经治疗的患者。这样的诊断可以仅仅基于被评估患者具有包含loh标记的基因组的癌细胞或至少部分地基于被评估患者具有包含loh标记的基因组癌细胞。例如,基于一种或多种肿瘤抑制基因(如,brca1/2、rad51)中的阳性loh标记状态和缺陷状态、癌症的家族史、或行为危险因素(如,吸烟)的存在的联合,已被确定具有包括loh标记存在的基因组的癌细胞的患者可被诊断为可能响应特定的癌症治疗方案。如本文中所述,已被确定具有包括loh标记存在的基因组的癌细胞的患者可被诊断为可能响应包括应用铂类化疗药物如顺氯氨铂、卡铂、奥沙利铂或吡铂,蒽环类如表阿霉素或阿霉素,拓扑异构酶i抑制剂如喜树碱、拓扑替康或伊立替康,parp抑制剂,放射治疗或它们的组合物,或任何前述的和任何抗癌制剂的组合物的特定癌症治疗方案。在某些实施例中,患者是未经治疗的患者。当一个临床医生或医疗专业人员或一组临床医生或医疗专业人员确定了特定的被评估的患者包含具有缺乏loh标记的基因组的癌细胞,该临床医生或医疗专业人员(或一组)可以将患者鉴定为具有包括loh标记缺乏的基因组的癌细胞。在一些实施例中,患者是未经治疗的患者。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将已确定的具有缺乏loh标记的基因组的癌细胞的患者诊断为可能具有功能性hdr的癌细胞。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将已确定的具有缺乏loh标记的基因组的癌细胞的患者诊断为癌细胞中不太可能具有hdr路径中一个或多个基因的基因突变。在某些情况下,一个临床医生或医疗专业人员或一组临床医生或医疗专业人员将带缺乏loh标记的基因组的癌细胞的患者或带覆盖整条染色体的loh区域数目没有增加的癌细胞的患者诊断为具有不太可能响应基于铂的化疗药物如顺氯氨铂、卡铂、奥沙利铂或吡铂、蒽环类如表阿霉素或阿霉素、拓扑异构酶i抑制剂如喜树碱、拓扑替康或伊立替康、parp抑制剂或放射治疗、和/或更可能响应与hdr无关联的药物如一种或多种紫杉制剂、生长因子或生长因子受体抑制剂、抗代谢物制剂的癌症治疗方案。在一些实施例中,患者是未经治疗的患者。如本文中所述,本文件还提供了执行癌症诊断分析的方法,分析患者核酸样品(如,基因组核酸样品或扩增的基因组核酸样品)来确定患者的癌细胞是否具有包含loh标记和/或覆盖整条染色体的loh区域数量增加的基因组。在一些实施例中,患者是未经治疗的患者。例如,一个或多个实验室技师或实验室专业人员可以在患者的癌细胞的基因组中检测loh标记的存在或缺乏,或在患者的癌细胞的基因组中检测覆盖整条染色体的loh区域数量是否增加。在某些情况下,一个或多个实验室技师或实验室专业人员可以通过(a)接收来自患者的癌细胞样品、接收来自患者的癌细胞的基因组核酸样品、或接收来自患者癌细胞的富集和/或扩增的基因组核酸样品以及(b)利用对接收的材料进行分析(如,基于snp阵列的分析或基于测序的分析)来检测文中描述的loh标记或覆盖整条染色体的loh区域是否数量增加,来检测患者的癌细胞的基因组中loh标记的存在或缺乏或覆盖整条染色体的数量增加的loh区域的存在或缺乏。在某些情况下,一个或多个实验室技师或实验室专业人员可以直接或间接接收来自临床医生或专业护理人员的被分析样品(如,来自患者的癌细胞样品、来自患者癌细胞的基因组核酸样品、或来自患者癌细胞的富集和/或扩增的基因组核酸样品)。在一些实施例中,患者是未经治疗的患者。一旦一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员检测了文中所述的loh标记的存在,实验室技师或实验室专业人员(一组)可将癌细胞具有loh标记的患者鉴定为带有阳性loh标记状态的癌细胞。例如,一个或多个实验室技师或实验室专业人员可通过关联阳性loh标记状态或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有已被检测具有loh标记的癌细胞的患者判定为含有具有阳性loh标记状态的癌细胞。在某些情况下,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阳性loh标记状态、hdr状态的潜在缺陷、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测具有loh标记的癌细胞的患者判定为含有hdr缺陷的癌细胞。这样的鉴定可仅仅基于检测loh标记的存在或至少部分基于检测loh标记的存在。例如,一个实验室技师或实验室专业人员可基于阳性loh标记状态和在测试实验室开展的其它的遗传和生化试验结果的组合来将含有被测定具有loh标记的癌细胞的患者判定为具有hdr潜在缺陷的癌细胞。在一些实施例中,患者是未经治疗的患者。在某些情况下,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阳性loh标记状态、hdr途径中一个或多个基因的基因突变的潜在存在、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测为具有loh标记的癌细胞的患者判定为具有潜在包含hdr途径中一个或多个基因的基因突变的癌细胞。这样的鉴定可仅仅基于检测loh标记的存在或至少部分基于检测loh标记的存在。例如,一个实验室技师或实验室专业人员可基于阳性loh标记状态和在测试实验室开展的其它的遗传和生化试验的组合来将含有被测定具有loh标记的癌细胞的患者识别于具有潜在包含hdr途径中一个或多个基因的基因突变的癌细胞。在一些实施例中,患者是未经治疗的患者。在某些情况下,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阳性loh标记状态、hdr状态的潜在缺陷、hdr途径中一个或多个基因缺陷状态的潜在存在、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测具有loh标记的癌细胞的患者判定为含有可能响应特定癌症治疗方案的癌细胞。这样的鉴定可仅仅基于检测loh标记的存在或至少部分基于检测loh标记的存在。例如,一个实验室技师或实验室专业人员可基于阳性loh标记状态和在测试实验室开展的其它的遗传和生化试验结果的组合来将含有被测定具有loh标记的癌细胞的患者识别于含有可能相应特定癌症治疗方案的癌细胞。在一些实施例中,患者是未经治疗的患者。一旦一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员检测了loh标记的缺乏,实验室技师或实验室专业人员(一组)可将患者(其癌细胞被检测缺乏loh标记)判定为含有具有阴性loh标记状态的癌细胞。例如,一个或多个实验室技师或实验室专业人员可通过关联阴性loh标记状态或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测缺乏loh标记的癌细胞的患者判定为含有具有阴性loh标记的癌细胞。在某些情况下,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阴性loh标记状态、潜在完整的hdr状态、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测缺乏loh标记的癌细胞的患者判定为含有具有潜在完整的hdr的癌细胞。在一些实施例中,患者是未经治疗的患者。在某些实施例中,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阴性loh标记状态、hdr途径中的基因缺乏基因突变、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测缺乏loh标记的癌细胞的患者判定为含有具有hdr途径潜在完整基因的癌细胞。在一些实施例中,患者是未经治疗的患者。在某些情况下,一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员可通过关联阴性loh标记状态、潜在完整的hdr状态、hdr途径中基因的基因突变的潜在缺乏、或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测缺乏loh标记的癌细胞的患者判定为含有不太可能响应一种特定治疗(如,铂类化疗药物如顺氯氨铂、卡铂、奥沙利铂或吡铂,蒽环类如表阿霉素或阿霉素,拓扑异构酶i抑制剂如喜树碱、拓扑替康或伊立替康,parp抑制剂如iniparib,olaparib或velapirib,或放射治疗)和/或更可能响应一种特定癌症治疗方案(如,包括与hdr不相关的癌症治疗制剂的癌症治疗方案)的癌细胞。在一些实施例中,患者是未经治疗的患者。一旦一个实验室技师或实验室专业人员或一组实验室技师或实验室专业人员检测了覆盖整条染色体的loh区域数量增加,实验室技师或实验室专业人员(一组)可将患者(其癌细胞被测定含有覆盖整条染色体的数量增加的loh区域)判定为可能含有具有完整的brca1、brca2和/或rad51c状态或完整的hdr途径的癌细胞。例如,一个或多个实验室技师或实验室专业人员可通过关联覆盖整条染色体的loh区域数量增加或已经开展的对应患者名字、病例、符号/数字标识符或其组合的诊断分析结果(或结果或结果的总结)将含有被检测具有覆盖整条染色体的loh区域数量增加的癌细胞的患者判定为可能含有具有完整的brca1和brca2状态的癌细胞。在一些实施例中,患者是未经治疗的患者。任何根据本发明的分析的结果将被传递给医师、遗传咨询师和/或患者(或其它有关各方如研究者),以可被传递或传送到任何上述方的可传送的形式。这种形式可以不同,并且可以是有形的或无形的。结果可被体现在描述性说明、图解、照片、图表、图像或任何其它视觉形式。例如,展示基因型或loh(或hrd状态)信息的图表或图解可被用来解释结果。该声明和视觉形式可被记录在有形介质如纸张、电脑可读介质如软盘、光盘、闪速存储器等,或无形介质如互联网或内联网上的电子邮件或网页形式的电子媒介。此外,结果还可被记录在声音形式和通过任何合适的介质(如,模拟或数字电缆线、光纤电缆等)用电话、传真、无线移动电话、互联网电话等被传输。因此,测试结果中的信息和数据可在世界上的任何地方被产生并被传送到不同的位置。作为一个说明性的例子,当实验是在美国境外进行,测试结果中的信息和数据可以被生成、以上述描述的传输形式传送,然后进口到美国。相应的,本发明还包括一种生产至少一个患者样品的loh标记信息的传送形式的方法。该方法包括步骤(1)根据本发明的方法检测loh标记;并且(2)以传输的形式具体化检测步骤的结果。该传输形式是这种方法的产物。在文中描述的本发明的一些实施例包含将根据本发明的loh标记(如,所述癌细胞中至少一对人类染色体上长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的总数,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5或更多百万碱基)与特定临床特征(如,brca1或brca2基因缺陷可能性的增加;hdr缺陷可能性的增加;响应包括dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、和/或parp抑制剂等的治疗方案的可能性的增加)相关联的步骤,如果数量大于一定的参考(或任选另一特征,如果该数目小于一定的参考)。在本文档中,无论被描述的这样的实施例,本发明的另外的实施例,除了或替代关联步骤,可能涉及以下步骤中的一个或两个:(a)断定患者具有至少部分基于loh标记存在或缺乏的临床特征;或(b)传达患者具有至少部分基于loh标记存在或缺乏的临床特征。以说明而非限制的方式,本文档描述的一个实施例是预测癌症患者响应包括dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、和/或parp抑制剂的癌症治疗方案的方法,所述方法包括:(1)在来自所述癌症患者的癌细胞中检测所述癌症患者的癌细胞中至少一对人类染色体上的长于第一长度的loh区域数量,该loh区域短于包含该loh区域的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5或更多百万碱基;并且(2)将所述大于参考数的总数与所述癌症患者将会响应所述癌症治疗方案的可能性的增加相关联。根据前面的规定,本实施例的描述应理解为是包括两个相关的实施例的描述,如,预测癌症患者响应包括dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、和/或parp抑制剂的癌症治疗方案的方法,所述方法包括:(1)在来自所述癌症患者的癌细胞中检测所述癌症患者的癌细胞中至少一对人类染色体上的长于第一长度的loh区域的数量,所述loh区域的长度短于包含该loh区域的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5或更多百万碱基;并且(2)(a)至少一部分基于大于参考数的总数,断定所述癌症患者响应癌症治疗方案的可能性增加;或(2)(b)至少一部分基于大于参考数的总数,传达所述癌症患者响应癌症治疗方案的可能性增加。在每个在本文档中描述的参与将特定试验或分析输出(如,大于参考数的loh区域的总数,等)与一些临床特征(如,响应特定的治疗、癌症特异性死亡、等)的一些可能性(如增加的、没增加的、下降的、等)相关联的,或附加地或可选择地断定或传达这样的至少一部分基于这样特定试验或分析输出的临床特征的实施例中,这样的关联、断定或上传可以包括至少一部分基于特定试验或分析输出,配置该临床特征发生的风险或可能性。在一些实施例中,这样的风险是事件或结果发生的概率百分比。在一些实施例中,患者被分配到风险组(如,低风险、中风险、高风险,等)。在一些实施例中,低风险是低于5%、10%、15%、20%、25%、30%、35%、40%、45%或50%的任何概率百分比。在一些实施例中,中等风险是指高于5%、10%、15%、20%、25%、30%、35%、40%、45%和50%并且低于15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%或75%的任何概率百分比。在一些实施例中,高风险是指高于25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或99%的任何概率百分比。在这里用到的,传递一条特定的信息意思是让这样的信息被他人知道或传送这样的信息到一个物体(如,电脑)。在本发明的一些方法中,传达了患者的预后或响应特定治疗的可能性。在一些实施例中,传达了用于达成这种预后或响应预测(如,根据本发明的loh标记,等)的信息。这种交流可以是听觉的(如,口头的)、视觉的(如,书写的)、电子的(如,从一个电脑系统传递到另一个的数据),等。在一些实施例中,传递癌症分类(如,预后、响应可能性、合适的治疗,等)包括生成传达癌症分类的报告。在一些实施例中,报告是纸张报告、听觉报告或电子报告。在一些实施例中,报告被显示和/或被存储在计算设备中(如,手持设备、台式电脑、智能设备、网站、等)。在一些实施例中,癌症分类被传递到医师那里(如,提供传递分类的报告给医师)。在一些实施例中,癌症分类被传递给患者(如,提供传递分类的报告给患者)。传递癌症分类还可以通过转移体现分类的信息(如,数据)至服务器并且允许中间人或终端用户使用这样的信息(如,通过观察显示在服务器的数据、通过下载从服务器转移到中间人或终端用户设备的一个或多个文件形式的信息,等)来完成。无论在什么情况下本发明的实施例包括断定一些事实(如,患者的预后或患者响应特定治疗方案的可能性),这可能包括在一些实施例中,断定这样的事实的计算机程序,典型的,在执行一种应用根据本发明的loh区域信息的算法之后。在文中描述的包括若干loh区域(如,loh指示区域)或这样loh区域总的叠加长度的每个实施例中,本发明包括涉及来源于、合并和/或至少某种程度上反应这样数量或长度的测试值或分值(如,hrd分值、loh分值、等)的相关实施例。换句话说,单纯的loh区域数量或长度不必用在本发明的各种方法、系统中;可以用源于这样数量或长度的测试值或分值。例如,本发明的一个实施例提供了治疗患者癌症的方法,包括:(1)在来自患者的样品中检测癌症患者癌细胞中至少一对人类染色体上的长于第一长度的loh区域的数量,所述的loh区域短于包含该loh区域的整条染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5或更多百万碱基;(2)提供来源于所述loh区域数量的测试值;(3)将所述测试值与一个或多个来源于参考人群(如,平均值、中值、百分位点、四分位点、五分位点、等)中的所述loh区域数量的参考值做比较;以及(4)(a)至少一部分基于所述比较步骤揭示所述测试值大于至少一种所述参考值(如,至少2-、3-、4-、5-、6-、7-、8-、9-或10-倍地大于;至少大1、2、3、4、5、6、7、8、9或10个标准差)而给予所述患者一种抗癌药,或推荐、开处方或开始包括化疗和/或合成致死药物的治疗方案;或(4)(b)至少一部分基于所述比较步骤揭示所述测试值大于至少一种所述参考值(如,至少2-、3-、4-、5-、6-、7-、8-、9-或10-倍地大于;至少大1、2、3、4、5、6、7、8、9或10个标准差)而给患者推荐、开处方或开始一种不包括化疗和/或合成致死药物的治疗方案。本发明包括,加上必要的修饰,相应的实施例,其中测试值或分值被用于判定患者的预后、患者响应特定治疗方案的可能性,患者或患者样本具有brca1、brca2、rad51c或hdr缺陷等等。图15展示了通过计算机系统(或包含计算机可执行指令的计算机程序(如,软件))利用此处描述的基因型数据鉴定loh位点或区域的示例性过程。如果两个等位基因a和b的可观察到的信号比是2到1,有两种可能性。第一种可能性是有50%正常细胞污染样品的具有loh的癌细胞中有等位基因b缺陷。第二种可能性是在没有被正常细胞污染的样品中没有loh但是有等位基因a的复制。该过程在方框1500处开始,其中下面的数据是由计算机系统收集的;(1)每个位点的两个等位基因的样品特异性标准化信号强度以及(2)基于对大量已知ascn谱的样品的分析来定义的方法特异性参数组合(具体针对不同的snp阵列和基于序列的方法)。如本文中所描述的,任何合适的试验如基于snp阵列试验或基于测序试验可被用来评估沿着染色体的位点的纯合性或杂合性。在某些情况下,包括信号检测器和电脑的系统可被用来收集关于多个位点(如,每个位点的两个等位基因的样品特异性标准化信号强度)的纯合或杂合性质的数据(如,荧光信号或测序结果)。在方框1510处,等位基因特异性拷贝数(ascn)在每个位点(如,每个snp)被重建。ascns是父系和母系等位基因的拷贝数。在方框1530处,似然函数被用来检测纯合的位点或纯合位点区域是否起因于loh。这在概念上类同于前面描述的旨在重建每个位点(如,snp)的总拷贝数(而不是ascn)的算法,见国际申请号pct/us2011/026098toabkevichetal。该似然函数可在所有位点的ascn处、正常组织的污染水平、在整条染色体上的平均总拷贝数、样品特异性噪声级处被最大化。在方框1540处,loh区域被确定作为一段具有一个ascns(父系或母系)为0的snps。在一些实施例中,电脑过程进一步包括调查或检测患者是否为未经治疗的患者的步骤。图3通过能检测loh谱的存在或缺失的计算机系统展示了代表性流程。该过程起始于方框300,其中关于沿着染色体的多数位点的纯合或杂合特征的数据通过计算系统被采集。如本文所描述,任何合适的试验如基于snp阵列试验或基于测序试验可被用来评估沿染色体的位点的纯合性或杂合性。在某些情况下,一个包括信号检测器和电脑的系统可被用来采集关于多个位点的纯合或杂合特性的数据(如,荧光信号或测序结果)。在方框310处,关于多个位点的纯合或杂合特性和每个基因位点的位置或空间关系的数据通过计算系统被评估来确定沿着染色体存在的任何loh区域的长度。在方框320处,关于被检测的loh区域数量以及每个被测loh区域的长度的数据通过计算系统被评估来确定具有(a)长度大于或等于预设mb数目(如,1.5mb)并且(b)长度小于包含该loh区域的染色体的整个长度的loh区域数量。或者,该计算机系统可以测定如前所述的总的或合并的loh长度。在方框330处,计算机系统将loh标记存在与否的指示信号输出格式化。一旦被格式化,该计算机系统可将这输出呈现给用户(如,实验室技术人员、临床医生或医疗专业人员)。如本文所描述,loh标记的存在与否可用来提供关于患者可能的hdr状态的显示,一种关于hdr途径中的基因突变可能存在或不存在的显示,和/或关于可能的癌症治疗方案的指示。图4是计算机设备1400和移动计算机设备1450一个实施例的图解,可以用于文中所描述的技术。计算设备1400意在代表各种形式的数字计算机,如笔记本电脑、台式机、工作站、个人数字助理、服务器、刀片服务器、大型机以及其它合适的计算机。计算设备1450意在代表各种形式的移动设备,如个人数字助理、细胞式电话、智能电话、以及其它类似的计算设备。在这里展示的组件,它们的连接和关系,以及它们的功能仅是示例性的,并不意味着限制在本文档中描述和/或要求保护的本发明的实施。计算设备1400包括处理器1402、存储器1404、一个存储设备1406、一个连接到存储器1404的高速接口1408和高速扩展端口1410,以及连接到低速总线1414的低速接口1415和存储设备1406。组件1402、1404、1406、1408、1410和1415中的每一个,使用各种总线互连,并且可能被安装到公共主板上或以其它合适的方式。处理器1402可以在计算设备1400内处理执行指令,包括存储在存储器1404或存储设备1406中的指令,来显示外部输入/输出设备上的图形用户界面的图形信息,如显示器1416结合到高速接口1408上。在其它实施中,多处理器和/或多条总线可沿着多个存储器和多种类型的存储器酌情被应用。并且,多个计算设备1400可被连接至每个提供必要操作部分(如,作为服务器,一组刀片服务器或多处理器系统)的设备上。存储器1404存储了计算设备1400中的信息。在一个实施中,存储器1404是易失存储单元或单位。在其它的实施中,存储器1404是非易失存储单元或单位。存储器1404可能还是其它形式的计算机可读介质,如磁盘或光盘。存储设备1406能够为计算设备1400提供大容量存储器。在一个实施中,存储设备1406可能是或包含计算机可读介质,如软盘设备、硬盘设备、光盘设备、或者磁带设备、闪存或其它相似的固态存储器设备、或一系列设备,包括在存储区域网络或其它配置中的设备。一种计算机程序产品可以有形地被体现在信息载体中。计算机程序产品可能还包括执行一种或多种那些文中描述的方法的指令。该信息载体是计算机或机器可读介质,如存储器1404、存储设备1406。处理器1402上的存储器、或传播的信号。高速控制器1808为计算设备1400管理带宽密集型操作,而低速控制器1415管理较低带宽密集型操作。功能的这样分配只是示例性的。在一个实施中,高速控制器1408结合到存储器1404、显示器1416(如,通过图形处理器或加速器)上、以及可能接收不同扩展卡(未显示)的高速扩展端口1410上。在这个实施中,低速控制器1415被结合到存储设备1406和低速扩展端口1414上。该可能包括各种通讯端口(如,ubs、蓝牙、以太网、或无线以太网)的低速扩展端口通过网络适配器可能被结合到一个或多个输入/输出设备,例如键盘、定点设备、扫描仪、光学阅读器、荧光信号检测器、或网络设备如交换机或路由器。计算设备1400可能以许多不同的形式被实现,如图所示。例如,可以被实施为标准服务器1420,或多次在一组这样的服务器中被实施。它还可以作为机架式服务器系统1424的一部分被实施。此外,它可能在个人电脑如手提电脑1422中被实施。二者择一地,来自计算设备1400的组件可能与移动设备(未显示)的其它组件合并,例如设备1450。每个这样的设备可能包含一个或多个计算设备1400、1450,以及可能由多个彼此连通的计算设备1400、1450组成的整个系统。计算设备1450包括其它组件(如,扫描仪、光学阅读器、荧光信号检测器)之间的处理器1452、存储器1464、输入/输出设备如显示器1454、通信接口1466、以及收发器1468。设备1450可能还装备有存储设备如微驱动器或其他设备来提供额外的存储。组件1450、1452、1454、1466、和1468中的每个可用各种各样的总线互相连接,并且若干组件可以其它适当的方式被安装在公共主板上。处理器1452可在计算设备1450中执行指令,包括存储在存储器1464中的指令。处理器可作为包括独立和多个模拟和数字处理器的芯片的芯片集被实现。处理器可提供,例如,设备1450的其它组件的协调,如用户界面的控制、设备1450运行的应用、以及设备1450运行的无线通信。处理器1452可能通过控制界面1458与用户沟通,并且通过显示界面1456结合到显示器1454。该显示器1454可能是,例如,一个tftlcd(薄膜晶体管液晶显示器)或一个oled(有机电致发光二极管)显示器、或其它合适的显示技术。显示器界面1456可能包括合适的电路驱动显示器1454来给用户呈现图解的和其它的信息。控制界面1458可能接收来自用户的指令并且转变它们提交给处理器1452。此外,外部接口1462可能被装备与处理器1452通信,以便能够使设备1450的区域通信接近其它设备。外部接口1462可能在一些实施例中提供,例如,有线通信,或在其它实施例中提供无线通信,并且多个接口也可以被使用。存储器1464在计算设备1450中存储信息。存储器1464可作为一个或多个计算机可读介质或介质、易失性存储器单元或单位、非易失性存储器单元或单位被实现。膨胀存储器1474也可以被提供并通过膨胀界面1472连接到设备1450,其可包括,例如,simm(单面接触内存模块)卡界面。这样的膨胀存储器1474可能为设备1450提供附加存储空间,或也可能为设备1450存储应用程序或其它信息。例如,膨胀存储器1474可能包括开展或补充本文描述的过程的指令,并且还可能包括安全信息。因此,例如,膨胀存储器1474可能作为设备1450的安全模块被提供,并且可能用允许安全使用设备1450的指令被编程。此外,安全应用程序可以经由simm卡连同其它信息被提供,如以不可非法侵入的方式将识别信息放在simm卡上。存储器可能包括,例如,如下所讨论的闪速存储器和/或nvram存储器。在一种实施方式中,一种计算机程序产品被有形地体现在一种信息载体中。该计算机程序产品包括指令,当其被执行的时候,完成那些本文中描述的一种或多种方法。信息载体是电脑或机器可读媒体,如存储器1464、膨胀存储器1474、处理器1452上的存储器、或可被接收的传播信号,例如,收发器1468或外部接口1462。设备1450可以通过通信接口1466进行无线通信,其中在必要时可以包括数字信号处理电路。通信接口1466可在不同的模式或方案下提供通信,如gsm语音电话、sms、ems、或mms消息传送、cdma、tdma、pdc、wcdma、cdma2000、或gpra,及其它。这样的通信可能发生,例如,通过射频收发器1468。此外,短距离通信可能会发生,例如利用蓝牙、wifi或其他这样的收发器(未显示)。此外,gps(全球定位系统)接收组件1470可能给设备1450提供额外的导航和位置相关无线数据,其可能通过在设备1450上运行的应用程序被适当的应用。设备1450还可使用音频编解码器1460可听见地通信,其可能从用户接收口头信息并将其转换为可使用的数字信息。音频编解码器1460可能同样的为用户产生可听见的声音,如通过扬声器,例如,设备1450的电话听筒。这样的声音可能包括来自语音电话的声音、可能包括被记录的声音(如,语音消息、音乐文件等)并且还可能包括通过在设备1450上操作的应用程序产生的声音。计算设备1450可以以许多不同的形式被实现,如图所示。例如,它可能作为蜂窝电话1480被实现。它也可能作为智能手机1482、个人数字助理或其它相似的移动设备的一部分被实现。本文中描述的系统和技术的各种各样的实施方式可在数字电子电路、集成电路、专门设计的asic(专用集成电路)、计算机硬件、固件、软件和/或它们的组合中被实现。这些各种各样的实施方式可包括在一个或多个在可编程系统(包括至少一个可编程处理器)上被执行和/或解释说明的电脑程序中的实施方式,其可以是专用或通用,被结合到接收来自存储系统、至少一个输入设备、和至少一个输出设备的数据和指令,并且传送数据和指令到存储系统、至少一个输入设备、和至少一个输出设备。这些电脑程序(也被称为程序、软件、软件应用程序或代码)包括可编程处理器的机器指令,并且可在高层次的程序和/或面向对象的编程语言、和/或装配/机器语言中被实现。在文中用到的,术语“机器可读媒体”和“电脑可读媒体”指的是任何用于给可编程处理器(包括接收机器指令作为机器可读信号的机器可读媒体)提供机器指令和/或数据的电脑程序产品、装置和/或设备(如,磁盘、光盘、存储器、可编程逻辑器件(pld))。术语“机器可读信号”指的是用于为可编程处理器提供机器指令和/或数据的任何信号。为了提供与用户的交互作用,本文中描述的系统和技术可在具有为用户和键盘和定点设备(如,鼠标或轨迹球)(由此用户可以为计算机提供输入)显示信息的显示设备(如,rct(阴极射线管)或lcd(液晶显示器)监控器)的电脑中被实施。其它种类的设备也可被用来提供与用户的交互作用;例如,提供给用户的反馈可以是任何形式的感官反馈(如,视觉反馈、听觉反馈或触觉反馈);并且来自用户的输入可以以任何形式包括声音、语音或触觉输入被接收。文中描述的系统和技术可在包括后端组件(如,作为数据服务器)或包括中间件组件(如,应用程序服务器)或包括前端组件(如,具有通过用户可以与本文中描述的系统和技术的实施相互作用的图形用户界面或web浏览器的客户端计算机)的计算系统、或这样的后端组件、中间件组件或前端组件的任何组合中被实施。该系统的组件可以通过数字数据通信(如,通信网络)的任何形式或介质被相互连接。通信网络的示例包括局域网(lan)、广域网(wan)和互联网。计算系统可包括客户端和服务器。客户端和服务器一般远离彼此并且通常通过通信网络交互。客户端和服务器的关系凭借在各自电脑上运行的并且具有彼此有关系的客户端-服务器的计算机程序上发生。在某些情况下,在文中提供的计算机系统可被配置为包括一个或多个样品分析仪。样品分析仪可被配置为产生多个关于癌细胞的至少一对人类染色体的基因组dna的信号。例如,样品分析仪可产生能够解析的信号用来鉴定沿着染色体的位点的纯合或杂合性质。在某些情况下,样品分析仪可以被配置为执行基于snp阵列的试验或基于测序试验的一个或多个步骤并且可以被配置为产生和/或捕获来自这样试验的信号。在某些情况下,本文中提供的计算机系统可以被配置为包括电脑设备。在这样的情况下,电脑设备可以被配置为接收来自样品分析仪的信号。该电脑设备可以包括计算机可执行指令或包含执行一个或多个文中描述的方法和步骤的电脑可执行指令的电脑程序(如,软件)。在某些情况下,这样的计算机可执行指令可以指导计算设备对来自样品分析仪、其它的计算设备、基于snp阵列试验或基于测序试验的信号进行分析。可以分析这样的信号来检测基因型、特定位点的纯合性、纯合区域、loh区域的数目,来检测loh区域的大小,检测具有特定大小或大小范围的loh区域的数目,检测样品是否有阳性的loh标记,检测至少一对人类染色体的指示loh区域的数目,检测brca1和/或brca2基因缺陷的可能性,检测hdr缺陷的可能性,检测癌症患者将响应特定癌症治疗方案(如,包括dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、parp抑制剂或它们的组合)的可能性,或检测这些项的组合。在某些情况下,本文中提供的计算系统可以包括计算机可执行指令或包含计算机可执行指令的计算机程序(如,软件)用于提供格式化的输出,所述输出提供关于loh区域数目、loh区域大小、具有特定大小或大小区域的loh区域的数目、是否一个样品是阳性的loh特性、至少一对人类染色体上指示loh区域的数目、brca1和/或brca2基因缺陷的可能性、hdr缺陷的可能性、癌症患者将响应特定癌症治疗方案(如,包括dns损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、parp抑制剂或它们的组合的方案)的可能性或这些项的组合的表示。在某些情况下,文中提供的计算系统可以包括计算机可执行指令或包含计算机可执行指令的计算机程序(如,软件)用于根据至少一部分基于loh标记存在与否或指示loh区域数量来判定特定患者的癌症治疗方案。在某些情况下,文中提供的计算系统可以包括被配置来处理样品(如,癌细胞)的预处理设备,这样可以开展基于snp阵列的试验或基于测序的试验。预处理设备的例子包括但不限于,被配置来从相对于非癌细胞中富集癌细胞细胞群的设备,被配置来裂解和/或提取基因组核酸的设备,以及被配置来富集样品的特定基因组dna片段的设备。本发明还提供了评估文中所描述的样品(如,癌细胞)的试剂盒。例如,提供了评估癌细胞loh标记的存在或检测至少一对人类染色体上的指示loh区域的数量的试剂盒。本文中提供的试剂盒可以包括snp探针(如,执行文中描述的基于snp阵列的试验)或引物(如,被设计用来通过基于测序的试验测序snp区域的引物)联合包含执行文中描述的一种或多种方法或步骤(如,检测具有特定大小或大小范围的loh区域的数量的电脑可执行指令)的计算机可执行指令的计算机程序产品。在某些情况下,文中提供的试剂盒可以包括能够与人类基因组dna的多态性区域杂交的至少500、1000、10,000、25,000或50,000个snp探针。在某些种情况下,文中提供的试剂盒可以包括能够测序人类基因组dna多态性区域的至少500、1000、10,000、25,000或50,000个引物。在某些情况下,文中提供的试剂盒可以包括开展基于snp阵列的试验或基于测序的试验的一个或多个其它成分。这样的其它成分的示例包括但不限于缓冲液、测序用核苷酸、酶(如,聚合酶),等。该文档还提供了利用文中提供的任何适当数目的材料制造执行文中描述的一种或多种方法或步骤的试剂盒。例如,本文档提供了利用一些snp探针(如,10,000-100,000的snp探针)和上文中提供的电脑程序产品制备评估癌细胞的loh标记存在的试剂盒。作为另一个例子,本文档提供了利用一些引物(如,10,000-100,000引物用于测序snp区域)和本文提供的计算机程序产品制备评估癌细胞的loh标记存在的试剂盒。本发明还包括下述实施方式:实施方式1.一种评估癌细胞或其基因组dna中loh的方法,其中所述方法包括:(a)在癌细胞或来源于其的基因组dna中检测所述癌细胞的至少一对人类染色体上的loh区域,其中所述至少一对人类染色体不是人类x/y性染色体对;并且(b)检测所述至少一对人类染色体上的长于第一长度但是短于包含所述loh区域的整条染色体长度的loh区域的总数,其中所述第一长度约1.5百万或更多百万碱基。实施方式2.一种预测癌细胞中brca1和brca2基因状态的方法,包括:在癌细胞中检测所述癌细胞的至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的总数,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;并且将大于参考数的所述总数与brca1或brca2基因缺陷的可能性的增加相关联。实施方式3.一种预测癌细胞中hrd状态的方法,包括:在癌细胞中检测所述癌细胞的至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的总数,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;并且将大于参考数的所述总数与hrd缺陷的可能性的增加相关联。实施方式4.一种预测癌症患者响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗,和/或parp抑制剂的癌症治疗方案的方法,所述方法包括:在来自所述癌症患者的癌细胞中检测所述癌症患者的癌细胞中至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的数目,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;并且将大于参考数的所述总数与所述癌症患者将会响应所述癌症治疗方案可能性的增加相关联。实施方式5.一种预测癌症患者响应治疗方案的方法,包括:在来自所述癌症患者的癌细胞中检测所述癌症患者癌细胞的至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的总数,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;并且将大于参考数的所述总数与所述癌症患者将不会响应包括紫杉醇或多烯紫杉醇的治疗方案可能性的增加相关联。实施方式6.一种治疗癌症的方法,包括:(a)在来自癌症患者的癌细胞或从其获得的基因组dna中检测癌细胞中至少一对人类染色体上的长于第一长度但是短于包含loh区域的整条染色体长度的loh区域的总数,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;并且(b)如果所述loh区域的总数大于参考数,给予所述癌症患者包含一种或多种选自由dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂组成的药物的癌症治疗方案。实施方式7.用一种或多种选自包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂和parp抑制剂的组合的药物制备对治疗已被鉴定为确定具有总数为5或更多的指示loh区域的癌细胞的患者的癌症有用的药剂。实施方式8.一种测定癌症患者癌细胞的loh状态的系统,包括:(a)配置用来产生多个关于所述癌细胞至少一对人类染色体的基因组dna的信号的样品分析仪,以及(b)基于所述多个信号,用来计算所述至少一对人类染色体上的指示loh区域数目的程序化的计算机子系统。实施方式9.根据实施方式8的系统,其中所述计算机子系统被编程将所述指示loh区域的数目与参考值做比较来确定(a)所述癌细胞中brca1和/或brca2基因缺陷的可能性,(b)所述癌细胞中hrd缺陷的可能性,或(c)所述癌症患者将会响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、或parp抑制剂的治疗方案的可能性。实施方式10.一种体现在计算机可读介质中的计算机程序产品,当在计算机上执行时,执行步骤包括:沿着除了人类x和y性染色体的一条或多条染色体检测任何loh区域的存在或缺乏,并且所述loh区域具有约1.5百万或更多百万碱基但是短于包含loh区域的整条染色体长度的长度;并且检测所述一对或多对染色体对中所述loh区域的总数。实施方式11.一种诊断试剂盒包括:能够与人类基因组dna的多个多态性区域杂交的至少500个寡核苷酸;以及实施方式10的计算机程序产品。实施方式12.能够与人类基因组dna的多个多态性区域杂交的多个寡核苷酸在制备对检测来自癌症患者的人类癌细胞的至少一对染色体中对指示loh区域总数有用的诊断试剂盒的用途,并用于检测:(a)所述癌细胞中brca1或brca2基因缺陷可能性的增加,(b)所述癌细胞中hrd缺陷可能性的增加,或(c)所述癌症患者将会响应包含dna损伤剂、蒽环类、拓扑异构酶i抑制剂、放射治疗、或parp抑制剂的治疗方案可能性的增加。实施方式13.实施方式1-6中任一项的方法,其中所述loh区域在至少2、5、10或21对人类染色体中被测定。实施方式14.实施方式1-6中任一项的方法,其中所述癌细胞是卵巢癌、乳腺癌或食管癌细胞。实施方式15.实施方式1-6中任一项的方法,其中所述loh区域的总数是9、15、20或更多。实施方式16.实施方式1-6中任一项的方法,其中所述第一长度约6、12、或15或更多百万碱基。实施方式17.实施方式1-6中任一项的方法,其中所述参考值是6、7、8、9、10、11、12、或13或更大。实施方式18.实施方式7或12的用途,其中所述指示loh区域在至少2、5、10或21对人类染色体中被测定。实施方式19.实施方式7或12的用途,其中所述癌细胞是卵巢癌、乳腺癌或食管癌细胞。实施方式20.实施方式7或12的用途,其中所述指示loh区域的总数是9、15、20或更多。实施方式21.实施方式7或12的用途,其中所述指示loh区域具有约6、12、或15或更多百万碱基的长度。实施方式22.实施方式8或9的系统,其中所述指示loh区域在至少2、5、10或21对人类染色体中被测定。实施方式23.实施方式8或9的系统,其中所述癌细胞是卵巢癌、乳腺癌或食管癌细胞。实施方式24.实施方式8或9的系统,其中所述指示loh区域的总数是9、15、20或更多。实施方式25.实施方式8或9的系统,其中所述指示loh区域具有约6、12、或15或更多百万碱基的长度。实施方式26.实施方式10的计算机程序产品,其中所述指示loh区域在至少2、5、10或21对人类染色体中被测定。实施方式27.实施方式10的计算机程序产品,其中所述癌细胞是卵巢癌、乳腺癌或食管癌细胞。实施方式28.实施方式10的计算机程序产品,其中所述指示loh区域的总数是9、15、20或更多。实施方式29.实施方式10的计算机程序产品,其中所述指示loh区域具有约6、12、或15或更多百万碱基的长度。实施方式30.实施方式1-6中任一项的方法,其中所述至少一对人类染色体不是17号人类染色体。实施方式31.实施方式7或12的用途,其中所述指示loh区域不在17号人类染色体上。实施方式32.实施方式8或9的系统,其中所述指示loh区域不在17号人类染色体上。实施方式33.实施方式10的计算机程序产品,其中所述指示loh区域不在17号人类染色体上。实施方式34.实施方式4或6的方法,其中所述dna损伤剂是顺铂、卡铂、奥沙利铂或吡铂;所述蒽环类是表阿霉素或阿霉素;所述拓扑异构酶i抑制剂是喜树碱、托普替康或伊立替康;或所述parp抑制剂是iniparib、olaparib或velapirib。实施方式35.实施方式12的用途,其中所述dna损伤剂是铂-基化疗药物,所述蒽环类是表阿霉素或阿霉素,所述拓扑异构酶i抑制剂是喜树碱、托普替康或伊立替康,或所述parp抑制剂是iniparib、olaparib或velapirib。实施方式36.实施方式9的系统,其中所述dna损伤剂是铂-基化疗药物,所述蒽环类是表阿霉素或阿霉素,所述拓扑异构酶i抑制剂是喜树碱、托普替康或伊立替康,或所述parp抑制剂是iniparib、olaparib或velapirib。实施方式37.实施方式10的计算机程序产品,其中所述dna损伤剂是铂-基化疗药物,所述蒽环类是表阿霉素或阿霉素,所述拓扑异构酶i抑制剂是喜树碱、托普替康或伊立替康,或所述parp抑制剂是iniparib、olaparib或velapirib。实施方式38.一种方法包括:(a)在癌细胞或来源于其的基因组dna中检测癌细胞的代表数量染色体对上的loh区域;并且(b)检测所述loh区域的数目和大小。实施方式39.实施方式38的方法,所述代表数量的人类染色体代表整个基因组。实施方式40.实施方式38的方法,进一步包含将特定大小的loh区域的数量增加与hrd缺陷的可能性的增加相关联。实施方式41.实施方式40的方法,其中所述特定大小是长于约1.5百万、2百万、2.5百万、3百万、4百万、5百万、6百万、7百万、8百万、9百万、10百万、11百万、12百万、13百万、14百万、15百万、16百万、17百万、18百万、19百万、20百万、25百万、30百万、35百万、40百万、45百万、50百万、75百万或100百万碱基并且少于包含loh区域的整条染色体的长度。实施方式42.实施方式40或41的方法,其中所述特定大小的6、7、8、9、10、11、12或13或更多数目的loh区域与hrd缺陷可能性的增加相互关联。实施方式43.一种确定患者预后的方法包括:(a)检测患者是否包括具有loh标记的癌细胞,其中癌症患者的癌细胞的至少一对人类染色体上的多于参考数的长于第一长度并且短于包含loh区域的整条染色体长度的loh区域的存在表明癌细胞具有loh标记,其中所述至少一对人类染色体不是人类x/y性染色体对,其中第一长度约1.5百万或更多的百万碱基,并且(b)(1)至少部分基于loh标记的存在,鉴别具有相对好的预后的患者,或者(b)(2)至少部分基于loh标记的不存在,鉴别具有相对不良预后的患者。实施方式44.一种包含选自由dna损伤剂、蒽环类、拓扑异构酶i抑制剂和prap抑制剂组成的组合的药物的组合物用于治疗选自由乳腺癌、卵巢癌、肝癌、食管癌、肺癌、头颈癌、前列腺癌、结肠癌、直肠癌、结直肠癌和胰腺癌的组合的癌症患者,所述患者的癌细胞具有多于参考数的在至少一对人类染色体上的loh区域,所述loh区域长于第一长度但是短于包含所述loh区域的整体染色体长度,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基。实施方式45.实施方式44的组合物,其中所述loh区域在至少2、5、10或21对人类染色体中被测定。实施方式46.实施方式44的组合物,其中所述loh区域的总数是9、15、20或更多。实施方式47.实施方式44的组合物,其中所述第一长度约6、12或15或更多百万碱基。实施方式48.实施方式44的组合物,其中所述参考值是6、7、8、9、10、11、12或13或更大。实施方式49.一种治疗患者癌症的方法,包括:在来自所述患者的样品中检测癌症患者的癌细胞的至少一对人类染色体上的长于第一长度并且短于包含loh区域的整条染色体长度的loh区域的数量表明癌细胞具有loh标记,其中所述至少一对人类染色体不是人类x/y性染色体对,其中所述第一长度约1.5百万或更多百万碱基;提供来自所述loh区域的数目的测试值;将检测值与来源于参考人群的所述loh区域数量的一种或多种参考值(如,平均值、中间值、百分位点、四分位数、五分位数、等)做比较;并且给予所述患者一种抗癌药物,或推荐或规定或启动包含化疗和/或合成致死剂的治疗方案,至少部分基于所述比较步骤,揭示了测试值是大于(如,至少2-、3-、4-、5-、6-、7-、8-、9-或10-倍大于;至少1、2、3、4、5、6、7、8、9或10标准差大于)至少一个所述的参考值;或推荐或规定或启动不包含化疗和/或合成致死剂的治疗方案,至少部分基于所述比较步骤揭示了测试值不大于(如,不大于2-、3-、4-、5-、6-、7-、8-、9-或10-倍,不大于1、2、3、4、5、6、7、8、9或10标准差)至少一个所述的参考值。实施方式50.实施方式49的方法,其中所述loh区域在至少2、5、10或21对人类染色体中被测定。实施方式51.实施方式49的方法,其中所述的loh区域的总数是9、15、20或更多。实施方式52.实施方式49的方法,其中所述第一长度约6百万、1千2百万或1千5百万或更多碱基。实施方式53.实施方式49的方法,其中所述参考值是6、7、8、9、10、11、12或13或更大。实施方式54.实施方式49的方法,其中所述化疗选自包含dna损伤剂、蒽环类。拓扑异构酶i抑制剂和/或所述综合致死剂是parp抑制剂药物的组合。实施方式55.实施方式49的方法,其中所述dna损伤剂是顺铂、卡铂、奥沙利铂或吡铂;所述蒽环类是表阿霉素或阿霉素;所述拓扑异构酶i抑制剂是喜树碱、托普替康或伊立替康;或所述parp抑制剂是iniparib、olaparib或velapirib。本发明将在下面的实施例中进一步被描述,这些实施例不限制权利要求中描述的本发明的范围。实施例实施例1–评估loh区域和hdr选用取自晚期卵巢癌患者的两组肿瘤。第一组的94例肿瘤(训练集)被用来获得候选标记,第二组的40例肿瘤(验证集)被用来验证标记。brca1和brca2基因的所有编码区域被测序来检测生殖细胞系和体细胞突变。测定brca1和brca2的mrna表达水平,并且开展affymetrixsnp微阵列分析。一种电脑程序被用来以源于微阵列数据的等位基因强度为基础重建loh标记状态。一种算法被开发并作为电脑程序被实施用于以基因型(如,snp基因型)数据为基础重建loh区域。该算法的一个点是首先重建每个位点(如,snp)的等位基因特异性拷贝数(ascn)。ascns是父系和母系等位基因拷贝的数量。一个loh区域接着被确定为具有一个ascns(父本或母本)为零的一串snps。该算法基于最大化似然函数,并在概念上类似于旨在重建每个位点(如,snp)的总拷贝数(而不是ascn)的如前所述的算法。见国际申请号pct/us2011/026098toabkevichetal.该似然函数在涵盖所有位点的ascn、良性组织的污染水平、涵盖整个基因组的平均总拷贝数、以及样品特异性噪声水平处被最大化。算法的输入数据包括(1)每个位点的所有等位基因的样品特异性标准化信号强度以及(2)基于分析大量已知ascn谱的样品来定义的试验特异性(特定于不同的snp阵列和基于序列的方法)参数集合。为了这个分析,将肿瘤定义为hdr缺陷型如果它们具有brca1和/或brca2基因的一种或多种有害突变或者具有baca1mrna低表达。为了这项分析,剩下的肿瘤被定义为可能是hdr非缺陷型。研究了loh区域长度的分布(图5)。用了三种类别的loh区域:(1)影响整条染色体的loh;(2)通常影响部分染色体臂或整条染色体臂的大的loh区域(大于约15mb);以及(3)多个短的loh区域(少于约15mb)。第二,只用训练集评估这三种类别中的一种的loh区域的数量与hdr缺陷之间可能的相关性。它被发现(1)短的loh区域的数量与hdr缺陷无显著相关性(p>0.05);(2)涵盖整条染色体的loh与hdr缺陷略有相关性(p=0.0011);并且(3)大的loh区域的数量与hdr缺陷显著性相关(1.9e-8)。更具体地说,发现所有hdr缺陷的肿瘤具有高数目(如,9或更多)的大loh区域,然而多数可能是hdr非缺陷的肿瘤具有小数目的大loh区域(图6-8)。这可能是因为这些hdr非缺陷的肿瘤由于其它遗传变异事实上是hdr缺陷,虽然不具有brca1和brca2突变和mrna低表达。除了大的loh区域的数量,这些区域的总长度也与hdr缺陷显著相关。这些结果已经用验证组证实:(1)短的loh区域的数量与hdr缺陷无显著性相关(p>0.05),(2)涵盖整条染色体的loh与hdr缺陷略有相关性(p=0.05);并且(3)大的loh区域的数量与hdr缺陷显著性相关(p=3.9e-6)。134例来自训练组和验证组的合并数据组的肿瘤被分为三组:(1)如果它们具有brca1和/或brca2基因的一个或多个有害性突变或者如果它们具有低brca1mrna表达即为brca缺陷;(2)如果它们具有9个或更多的大loh区域(大于15mb但是少于整条染色体的长度)即为hdr缺陷/brca完整;(3)如果它们具有少于9个的大loh区域(大于15mb但是少于整条染色体的长度)即为hdr完整。这一分析结果呈现在表9。它显示了在卵巢癌中高频率的brca缺陷以及非因brca基因缺陷的hdr缺陷。图10展示了不同类型的癌细胞系中大loh区域(大于15mb但是少于整条染色体的长度)的分布。圆圈的大小与具有这样的loh区域的样品数目成比例。hdr缺陷的频率(具有至少9个如此的大loh区域的细胞系)在乳腺癌和食管癌细胞系中是最高的。结肠癌细胞系中没有hdr缺陷。验证了以前的卵巢肿瘤研究结果,所有的brca缺陷细胞系被发现也是hdr缺陷的。图11显示了公开的肺癌数据集(来自基因表达综合数据库的gse19399)中大loh区域(大于15mb但是少于整条染色体的长度)的分布。还发现hdr缺陷的频率(定义为具有至少9个大的loh区域)在肺肿瘤中是非常高的(39%)。图12总结了不同肿瘤以及细胞系的分析结果。具有至少9个大loh区域(大于15mb但是少于整条染色体的长度)的样品在所有样品中的比例定义为hdr缺陷的频率,很多肿瘤和细胞系中的这种hdr缺陷频率被提供于此。这种频率在卵巢肿瘤中高达50%,但在脑和结肠细胞系中没有观察到。似乎hdr缺陷在大多数癌症中起非常重要的角色。实施例2–化疗毒性反应为化疗毒性实验作准备,所有的细胞系在75cm2组织培养瓶(vwrinternational,inc.cat#353136)以及被推荐的生长介质中在37℃和5%co2条件下生长。在开展每个实验之前,每个细胞系被胰蛋白酶化(invitrogencorporationcat#25200-056)、计数并且将2500个细胞或5000个细胞于100μl改良的rpmi1640(invitrogencorporationcat#12633-020),3%fbs,1%青霉素/链霉素(invitrogencorporationcat#15140-122)中接种于有洁净底面的96孔聚苯乙烯微孔板(perkinelmercat#6005181)的2-12排的每孔中,剩余的第一排每孔只加100μl培养基。接种了细胞的培养板在37℃和5%co2孵育过夜。制备了两种不同最终药物浓度的工作贮存液。在药物稳定情况下,需要100%dmso,用改良的rpmi1600作为最高浓度的稀释剂。低浓度工作贮存液中所用的改良的rpmi1600加预先测定的dmso的量与高浓度工作贮存液中的总dmso相等,最低浓度需要用最多60%的dmso。这样做是为了保持每孔中dmso的浓度相等并且预防由于dmso引起的非特异性细胞死亡。96孔的薄壁pcr循环板(robbinsscientificcat#1055-00-0)的第12列的a-d行加入了较低的两种药物浓度,而同一板的第12列的e-h行加入了较高的浓度。从第12列到第3列以递减的方式做了一系列1:2或1:3的稀释,留下第1列和第2列作为无细胞/无药物和无药物的对照。这样每个药物浓度有四个重复。一旦完成稀释,从稀释板转移5μl至接种细胞板的相应孔中。接受药物的板接着在37℃和5%co2条件下孵育3天或6天。一个3天或6天的给药方案之后,每个板的每孔根据atplite试验方案运行atplite试验(perkinelmercat#6016941)。然后在fusion机器上读取荧光并保存为.csv文件。对于每个细胞系和药物浓度,算出无药物对照的四个重复的平均值并且除以100来创建用来计算标准化生存率的标准化因子。无药物对照的标准化存活率是100%。算出细胞加药物孔的四个重复的平均值并且除以每个药物浓度的标准化因子。每个药物浓度的存活率,起始浓度等于0,用于利用专用软件计算ic50。图13显示了响应化疗的乳腺癌和卵巢癌细胞系。y-轴表示当暴露于29例乳腺癌细胞系后不同化疗药物(喜树碱、以及铂化合物(奥沙利铂、顺铂、或卡铂)或蒽醌类(表阿霉素和阿霉素)的平均结果)的log10(ic50)值以及当暴露于27例卵巢癌细胞系后紫杉醇的log10(ic50)值。x-轴指示这些细胞系的长于15mb并且短于整条染色体的大loh区域的数量。虚线将阙值设置在9。图14是来自于图13的显示当暴露于29例乳腺癌细胞系后对用铂化合物(奥沙利铂、顺铂和卡铂)治疗的响应者和非响应者的特异性和敏感性的图表的一个版本。虚线将大于15mb并且短于整条染色体的大loh阙值数目设置在9。实线将细胞系划分为响应者和非响应者。实施例3–hr缺陷试验的进一步验证材料和方法卵巢肿瘤样品用了三个独立的人类卵巢癌试验群组。1、152例未经选择的卵巢癌样品。2、53例高级别卵巢浆液性肿瘤。3、完整信息可见的来自435例卵巢浆液性肿瘤样品的公开数据在2011年10月31日从癌症基因组图谱(tcga)网络网站下载。所有群组的病人根据机构审查委员会(irb)批准的协议获得。患者和肿瘤特性见表2。不同数量的样品用于下述试验(表3)。表2.患者和癌症特性表3每个实验中的样品数细胞系分析了67例癌细胞系(29例卵巢癌、34例乳腺癌、3例结肠癌、1例胰腺癌)。三例乳腺癌细胞系从德国微生物保存中心(braunschweig,germany)获得。结肠癌、胰腺癌以及剩余的乳腺癌细胞系从美国模式培养物研究所(manassas,va)获得。癌细胞系在rpmi+10%fbs+1%青霉素/链霉素的培养基中在37°c的t75培养瓶中生长直至细胞密度约为5x106。例外是需要非标准培养基、l-谷氨酰胺或胰岛素的细胞系。生长于悬浮液中的细胞用1.5ml的离心管在1700rpm下离心5分钟,弃上清。抽吸去除单层生长的细胞中的培养基,用pbs洗涤,并加入胰蛋白酶溶液。细胞分离后收集在培养液中,转移至1.5ml微量离心管中并且在1700rpm下离心5分钟。弃上清,将分离的细胞重悬于200μlpbs。从冰冻肿瘤以及细胞系中提取基因组dna和总rna切取10μm的冰冻切片并巨切。组织加入qiazol裂解试剂后匀浆(tissueruptor(qiagen)),接着用qiagenmirnaeasy迷你试剂盒根据厂商手册分离rna。用qiaampdnaminikit试剂盒(qiagen)根据厂商手册在56℃孵育裂解一晚并用rnasea处理来分离dna。brca1和brca2测序brca1和brca2测序根据hennessyetal.,2010描述的方法进行。鉴定的突变仅包括,基于前面描述的标准,在分析中是否被归类于有害的或疑似有害的(beaudetandtsui,1993)突变。启动子甲基化qpcr试验根据生产商介绍的方案用methyl-profilerdna甲基化pcr分析系统(sabiosciences)来量化甲基化水平。分别用dna甲基化敏感的和甲基化依赖的限制性内切酶选择性的酶切未甲基化的或甲基化的基因组dna。用侧面连接兴趣区的引物通过实时pcr量化酶切后的dna。差异甲基化的dna的相对浓度通过将每个酶切的量与模拟酶切的量进行比较来测定。brca1启动子甲基化测序分试验50-300ngdna在60℃孵育约5小时,并在硫酸氢盐存在的酸性环境下短暂升温至95℃。孵育后,将反应绑定到吸附柱并在碱性条件下洗涤去除硫酸氢盐,转化的dna在15μl处被洗脱。引物的小写字母区域特定于被扩增的基因组区域。引物的大写字母区域对应于454钛化学尾巴以及4bp条形码(区域特定碱基前的最后4个碱基)。通过在多重组合中合并正反向引物,在一个单序列反应中复用多达100个样品是可能的。brca1和细胞周期进程特性表达试验根据生产商手册用扩增级脱氧核糖核酸酶i(sigma-aldrichinc.)处理rna并延长孵育时间至30分钟。用大容量cdna反转录试剂盒(appliedbiosystems,fostercity,ca)根据生产商的说明进行反转录。用taqmanpreampmastermix试剂盒(appliedbiosystems)方案在5μl反应容积内独立运行复制前置放大。在细胞周期基因试验中分别运行前置放大复制8个循环和18个循环。只在brca1试验中运行三个前置放大复制18个循环。将扩增后的产物按1:5稀释于低-edtatris-edta(te)中。实施定量聚合酶链式反应(qpcr)并用基因表达m48动态试验(fluidigm,southsanfrancisco,ca)根据生产商的方案评估。用比较循环阙值(ct)方法计算相对基因表达。通过所有复制中共有的复制上的基因的平均值来集中来自不同循环数的扩增前的cts。首先用管家基因的平均cts标准化结果值接着用来自第一队列人群的所有样品的每个试验的标准化cts的平均值来获得δδct。计算ccp得分以及相对brac1表达,分别作为细胞循环基因以及brca1试验的δδcts负值的平均值。brca1表达缺失的样品的鉴定:ccp表达和brca1表达反相关的样品被定义为brca1缺陷。在大量的卵巢癌样品(n=300)中用稳健线性回归定义鉴定具有异常brca1表达的患者的阙值。brca1表达用迭代重新加权最小二乘(iwls)回归ccp得分。在低端99%区间范围外的点被认为是异常的。该方法在国际申请号pct/us2011/054369totimmsetal中有更详细的描述。affymetrix500k基因芯片试验用affymetrix基因芯片绘图nspi或styi试验试剂盒来生成生物素化的dna用于affymetrixmapping500knspi或styi微阵列杂交(分别制备每个试验)。用nspi或styi限制性内切酶消化基因组dna(250ng)并在末端具有t4dna连接酶的限制性片段中加入接头。用clontech钛taq酶pcr扩增接头修饰的样品,生成平均大小在200-1100bp之间的扩增产物。用clontechdna扩增清除试剂盒纯化扩增产物。用affymetrix破碎试剂破碎90μg纯化的dna。用基因芯片dna标记试剂完成片段化样品的生物素标记。生物素标记的dna在nspi或styiaffymetrix微阵列上于affymetrix旋转炉中在49℃杂交16-18小时。杂交后,在自动affymetrix流体站根据生产商的手册实施探针阵列清洗和染色程序。扫描微阵列,并用affymetrix基因芯片扫描仪3000采集原始数据。snp微阵列数据的cn和loh分析设计算法确定每个snp位置的最可能的等位基因特异性的拷贝数。对应的可能性明确的考虑到非癌基质细胞dna对癌症dna样品的污染。cn分析的类似算法在国际专利申请号pct/us2011/026098toabkevichetal.(publicationno.wo/2011/106541)中被详细的描述。在本文中所使用的算法有两个版本,一个用来分析内部产生的affymetrix500k基因芯片试验数据,另一个是用来分析从tcga站点(http://tcga-data.nci.nih.gov/tcga/dataaccessmatrix.htm?diseasetype=ov)下载的基因组widesnp6affymetrix试验数据。后面的试验中,除了snp探针,包括交叉人类基因组的非多态性位点的一些探针。这些探针对cn分析来说是大信息量的,但是对loh分析没有直接的信息功能。统计分析在本文中,除非另有规定,用kolmogorov-smirnov试验计算p-值。结果hr缺陷肿瘤如果具有brca1或brca2的生殖系或体细胞突变,或brca1甲基化或低brca1mrna表达量,该肿瘤样品被认为是hr缺陷的。来自第一队列人群的152例样品中的31例,来自第二队列人群的53例中的14例以及来自第三队列人群(其中两个在进一步的分析中被被排除,见下文)的435例中的83例是brca1和/或brca2突变的携带者。突变总结在表4中。表4.在研究队列人群中检测到的brca1、brca2以及rad51c缺陷1–这些突变中的两个被排除在分析之外因为brca2基因的一个拷贝保持完整。测量了brca1和brca2的启动子cpg岛的甲基化程度。在多个样品中观察到了brca1甲基化,但没有观察到brca2的甲基化。因为高水平的brca1启动子甲基化,来自第一队列人群的126例样品中的11例,来自第二队列人群的34例样品中的3例,以及来自第三队列人群的435例样品中的64例被定义为hr缺陷。除了第三队列人群中的一例样品,在这些样品中的任何一个中并未观察到有害的brca1或brca2突变。低brca1或brca2的mrna表达可能还会导致hr缺陷,并且可能是由于除启动子甲基化的机制导致的。测定了来自第一队列人群的137例样品和来自第二队列人群的53例样品的brca1和brca2的表达水平。20例样品中的brca1表达异常的低。只有具有异常低brca1表达的五例样品没有标记为hr缺陷,这归因于brca1启动子甲基化。没有发现异常低表达的brca2。brca1或brca2的单个完整拷贝对功能化来说是必须的。对所有的brca1缺陷样品来说,brca1基因包含在loh区域中。此外,在所有的样品中,仅有两个brca2缺陷样品的loh区域中观察到了brca2基因。在我们的分析中,这两个brca2缺陷样品不认为是hr缺陷。loh区域的长度分布一开始的假设是具有不同长度的loh的区域可能通过不同的途径出现在癌症基因组中,loh和hr缺陷之间的这样的关联可能取决于loh区域的长度。loh区域的长度分布在染色体臂长度上做出了调整,在这上面观察到的这些loh区域展现在图16中。13、14、15和22号染色体被排除,这是因为对这些染色体的p臂来说snps是不适用的。在这些分布中观察到了三个明显的特征。首先,有很多短的loh区域。其次,有一个loh区域的长平尾,相当于单个染色体臂的长度。少量的loh区域覆盖多于一条染色体臂但是少于整条染色体。最后,有一个对应于覆盖整条染色体的loh的高峰。所观察到的分布与得到的cn变化(beroukhimetal.2010)的相似分布有着非常大的差异,这表明cn变化和loh区域可能是通过不同的机制引起的。样品中hr缺陷和loh的相关性样品中的第一队列人群被用作“发现”组人群。17号染色体上的loh区域被排除在分析之外,这是因为在几乎所有样品中观察到了该条染色体上的loh,这可能是因为对卵巢癌的进展重要的基因都在这条染色体上。我们检查了hr缺陷和短的loh区域(<15mb)数量、长的loh区域(>15mb但是短于整条染色体)的数量、以及覆盖整条染色体的loh区域的数量之间的相关性。尽管发现15mb是一般的首选,但是各种各样的不同的loh区域长度的阈值可用,并且这些阈值对检测hr缺陷的影响体现在图19及与之附随的讨论中。短的loh区域的数量与hr缺陷之间没有显著相关性。覆盖整条染色体的loh区域的数量在具有完整brca1或brca2的肿瘤中明显较大(p=4x10-5)。长的loh区域的数量(在后面的示例3以及在整个文档中被称为“hrd分值”)在具有缺陷的brca1或brca2的肿瘤中明显较高(p=9x10-11)(图17a)。第二队列人群和第三队列人群用于验证从第一队列人群获得的结果。hr缺陷和覆盖整条染色体的loh区域的数量之间的相关性没有在第二队列人群中验证,这可能是由于低样本数,但是在第三队列人群的具有完整的brca1和brca2的肿瘤中是明显较大的(p=3x10-11)。在具有完整的brca1和brca2的卵巢肿瘤之间直接降低的hrd分值(图17b和17c)的这两组队列人群中都观察到hrd分值和hr缺陷之间有非常显著的相关性(分别为p=2x10-7和p=9x10-30)。rad51c以及其它hr途径基因的变异现有数据表明,brca1和brca2是负责卵巢癌hr缺陷的主要基因。然而,其它基因对此可能也非常重要,例如,rad51c(meindletal.,2010)和rad51d(lovedayetal.,2011)最近被认为作为卵巢癌的易感基因。在第一队列人群中,测量了参与hr途径的8个其它基因的启动子cpg岛的甲基化程度(图5)。只有rad51c具有高水平的启动子甲基化(89例样品中的3例)。第三队列人群的435例样品中的11例具有rad51c启动子甲基化。来自两组队列人群的rad51c甲基化阳性的所有样品在rad51c位点是纯合的,这归因于loh。为了检验hrd分值在具有rad51c启动子甲基化的样品中是否升高,来自两组队列人群的这些样品与不具有rad51c甲基化的brca完整样品做了比较。这符合我们对brca1和brca2基因的观察,hrd分值在具有rad51c甲基化的样品中明显更高。表5.用到的启动子甲基化分析(sabiosciences).基因符号说明分析目录idmdc1炎症介质或dna损伤检验点1meph08721-2aparp1多聚(腺苷二磷酸-核糖)聚合酶1meph02379-2abrca1乳腺癌1,早发型meph28472-1abrca2乳腺癌2,早发型meph28473-1arad50rad50同系物meph28350-1arad51crad51同系物cmeph22389-1apalb2brca2合作者和定位信标meph28516-1achek2chk2检验点同系物meph28264-1aatm突变的共济失调毛细血管扩张症meph28470-1arad51rad51同系物meph19071-2a第三队列人群中,有报道hr途径基因的有害突变和甲基化(tcga,2011)。检查了突变,并且分析仅限于很可能有害的缺陷(如,无义突变和移码突变),发现了6个基因中(atm、atr、fanca、fancd2、fancm和palb2)总共有8个有害突变。额外的5例样品具有hr途径基因甲基化。只在13例样品中的1例(fancm无义突变)中测定了第二等位基因缺失。因为两个等位基因同时失活对肿瘤抑制因子的功能丧失是必须的,这些13例样品中的大多数被预测具有完整的hr。hrd分值在大多数的这些样品中没有升高。总合数据分析hrd分值与所有三组队列人群的hr缺陷(定义为brca1、brca2、或rad51c缺陷)之间的相关性列于图17d。可见高显著相关性(p=2x10-54)。一个重要的问题是hrd分值的分布是否与hr缺陷一样归咎于不同基因位点。为了回答这个问题,分别分析了brca1、brca2和rad51c缺陷肿瘤的hrd分值分布(图21)。可观察到显著的不同(p=7x10-5),brca1缺陷样品(16.1;sd=4.3)比brca2缺陷样品具有更高的平均hrd分值(13.0;sd=3.9)。brca1或brca2和rad51c之间的hrd分值差异不显著(14.5;sd=5.1)。正常组织可以来自第一二组队列人群的一部分样品以及第三队列人群的所有样品,这些用于决确定brca1和brca2的突变是否为生殖系的或体细胞系的。体细胞的和生殖系的brca1或brca2缺陷的hrd分值的分布无显著性差异(图20)。brca1和brca2缺陷细胞系的hrd分值未经选择的乳腺(n=34)和卵巢(n=29)细胞系从多个来源获得;另外分析了来自nci60的具有公开的brca1和brca2状态的3例结肠和1例胰细胞系。在这些67例细胞系中,7例携带了纯合的有害突变或具有brca1启动子甲基化,2例具有纯合子突变但有明显的功能恢复,并且6例携带杂合子突变。图18a显示了这些三组中的突变体以及野生型样品的hrd分值的分布。野生型卵巢肿瘤和野生型癌细胞系的hrd分值的分布没有显著差异。具有杂合突变的癌细胞系之间的hrd分值分布与野生型癌细胞系的hrd分值的分布是类似的,大概是因为细胞只有当brca1和brca2的两个拷贝为非功能性时变为hr缺陷。在具有brca1或brca2的两个拷贝功能丢失的癌细胞系中观察到了较高的hrd分值,这与在具有brca1、brca2或rad51c缺陷基因的卵巢肿瘤中所观察到得hrd分值类似。hrd分值在具有brca1和brca2突变回复的癌细胞系中也很高,这支持了原来的假设,也就是hr缺陷导致了loh的不可逆变化。hrd分值分布在野生型或杂合突变的细胞系中的差异,以及hrd分值分布在具有纯合突变(有或没有回复)或brca1启动子甲基化的细胞系中的差异是高度显著的(p=10-5)。重要的是,从资料组中排除卵巢癌细胞系后,hrd分值与brca1和brca2缺陷之间有显著相关性(p=0.01),这表明hrd分值与hr缺陷之间的相关性并不限于卵巢癌。hr缺陷与总生存数(os)和无进展生存率(pfs)之间的相关性对具有较高hrd分值的患者的高生存率的第三队列人群来说,在pfs(p=0.03)和os(p=6x10-5)之间观察到了显著的相关性(图18b)。用cox模型计算p-值。结果与以往报告显示brca1和brca2的生殖系突变与卵巢癌的更好的预后相关的数据(rubinetal.,1996,boydetal.,2000;cassetal.,2003;tanetal.,2008,hennessyetal.,2010,)一致并有延伸。讨论hrd分值在两个独立的卵巢癌数据集中被验证,同时也反映了导致乳腺和胰腺细胞系hr缺陷的突变。表6.brca1和brca2缺陷型和完整型肿瘤的hrd分值平均值以及相对应的p值长度大于15mb但是小于整条染色体的loh的这一中间组与缺陷型hr基因高度正相关表明大多数,如果不是所有的,这种类型的loh的存在是因为它包括双链dna断裂作为其起源的一部分并且需要hr修复。相反,整条染色体水平上的loh在hr缺陷的肿瘤中有着显著的低频率。一种可能的解释是,整条染色体水平上的loh通过不涉及双链dna断裂的替代竞争机制起源。除了brca1和brca2缺陷,卵巢肿瘤中观察到了rad51c启动子甲基化。在两个数据集中,高的hrd分值与rad51c缺陷显著相关。在这三种数据集中只确证了一个额外的hr缺陷肿瘤,具有loh的fancm的无义突变导致了第二等位基因的缺失。与fancm突变(8)相关联的hrd分值在具有升高的样品hrd分值的正常分布范围内。在具有明显完整brca1、brca2和rad51c的肿瘤中,样品中的大多数具有升高的hrd分值。一种可能的解释是在许多这些样品的hr途径的其它基因中有大幅率的缺陷。另一种解释是正常组织对肿瘤的污染使缺陷检查变得复杂化。数据表明相比较其它试验分析,hrd分值对污染不太敏感,未检测到的缺陷可能解释那些具有升高的hrd分值的样品的显著分数(见补充结果)。发表的研究表明恢复brca2功能的二次反向突变在brca2突变细胞系暴露于铂试剂后可产生(sakaietal.,2009;sakaietal.,2008;edwardsetal.,2008)。norquistetal.,(2011)观察到大约28%的复发肿瘤具有恢复brca功能的次级突变。反向突变主要出现在之前暴露于铂试剂并且被预测为耐铂的个体中。hrd分值起因于发生在肿瘤基因组的累计缺陷。基于dna的hr缺陷标记有可能与hr缺陷有强的相关性,因为它们在功能上相联。因此,hrd分值是hr缺陷非常有力的衡量指标。然而,它的持久性表明该分值对反向突变可能不太敏感,本研究中没有治疗过的肿瘤样品,然而从细胞系中得到的数据与该假设一致。检测不出反向突变将会导致假阳性,这有可能影响非常少的肿瘤的新辅助疗法或辅助疗法(norquistetal.,2011),这个问题的严重性比假阴性小,因为假阴性可能将个体误鉴定为非应答者。高hrd分值与hr缺陷高度相关,该分值可用于判定对响应dna损伤剂和parp抑制剂(在其它试剂之中)有高可能性的患者。这样的试验在乳腺癌和卵巢癌中有明显的临床应用,并且可用于扩展parpi和铂盐在其它没有很好表征hr缺陷的癌症上的应用。实施例4–hr缺陷试验的进一步验证材料和方法在本示例中被分析的患者组包括56例乳腺癌患者,所有人要么是阳性的brca突变要么是具有三阴性乳腺癌(大多数是tnbc)。包括i-iii期(大多数是ii或iii期)。患者接受了6个循环的新辅助疗法吉西他滨+iniparib+卡铂。治疗后测量相对较低的残留肿瘤负担作为响应。分析了56例新鲜冷冻的乳腺肿瘤样品。污染度的中间值是60%。9例样品的污染度至少为90%。这些肿瘤中的11例是brca1有害突变的携带者,三例是brca2有害突变的携带者。在所有的这些肿瘤的缺陷基因中有loh。brca1有害突变携带者中的一例还携带了brca2有害突变。然而这些样品的brca2基因没有loh。从响应治疗的患者(残存肿瘤负担是0或1)中获得30例样品。他们中的13例是brca1/2缺陷的。从非响应者(残存肿瘤负担是2或3)中获得26例样品。他们中的1例为brca1缺陷的。经affymetrix用affymetrixmip进行基因型分析(如u.s.pat.no.6,858,412;u.s.专利申请公布说明书no.us20060234264;hardenboletal.,naturebiotechnology(2003)21:673-678;wangetal.,bmcmedgenomics(2009)2:8所描述,其中每一个在此通过引用的方式全文并入)。hrd分值的计算如前所述。结果响应者的平均hrd分值是16.5。brca1/2完整的和brca1/2缺陷的响应者的平均hrd分值是一样的。非响应者的平均hrd分值是11.4。brca1/2完整的非响应者的平均hrd分值是11.6,brca1缺陷的非响应者的平均hrd分值是8。根据治疗响应与hrd分值之间相关性的曼-惠特尼u检验的p-值是0.004。即使brca1/2缺陷样品被排除,治疗响应与hrd分值之间相关性仍然显著(p-值=0.02)。在残存肿瘤负担为0和1的样品中hrd分值的差异不显著。类似的,剩余癌症负担为2和3的样品中hrd分值的差异不显著。治疗响应与临床参数(临床分期、临床级别)之间的相关性不显著。其它实施例应当理解的是,虽然本发明结合其详细说明进行了描述,前面的描述旨在说明而不是限制本发明的范围,这是由所附的权利要求的范围限定。其它的方面、优点和修改在以下权利要求的范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1