一种HuR蛋白可识别的RNA片段及在HuR活性检测中的应用的制作方法
本发明涉及分子生物学领域,更特别地,涉及一种hur蛋白可识别的rna片段及其在检测细胞内hur转录后调控活性中的应用。
背景技术:
人抗原r(humanantigenr,hur)属于rna结合蛋白中胚胎致死异常视觉(embryoniclethalabnormalvision,elav)家族,elav家族包括4个成员:hub、huc、hud及hur,前3个成员主要在神经组织和生殖器官中表达,并与神经发育有关。近年来研究表明,hur在靶基因转录后调控中发挥稳定mrna和促进翻译的作用。多项实验证实:hur结合的靶因子多是癌基因、抑癌基因及肿瘤发生的调控因子,因而hur与多种肿瘤的发生、发展及预后密切相关。hur与人乳腺癌、结直肠癌、宫颈癌、卵巢癌和胃癌的发生、侵袭及转移有关,可能是影响肿瘤发生、发展及预后的重要因素。
hur是一种mrna结合蛋白,其活性形式能够与mrna的3’-utr结合,并通过该方式来调控mrna的稳定性和翻译效率。因此,针对hur的研究需要检测hur的活性形式。
然而,目前检测内源性hur仍然依靠的是westernblot技术,虽然灵敏度高,但检测到的只是hur蛋白含量的差异,并不能直接体现其激活水平的改变。
因此,需要设计一种新的用于检测细胞内hur转录后调控活性的方法。
技术实现要素:
发明人通过研究发现,在hur蛋白上有3个rna识别模序(rrms),可以与ares特定位点结合。rrm1和rrm2与富含u/a拷贝序列结合。rrm3与poly(a)尾结合,帮助维持rna蛋白复合物的稳定。通过分析,发明人发现,将seqidno:1所示的序列编码的rna序列接在mrna3’端时,hur蛋白可有效结合该mrna的3’区,激活该mrna翻译并提高mrna的稳定性。
基于以上研究,本发明提供了一种hur蛋白可识别的rna片段,其由seqidno:1所示的dna序列转录得到。
本发明还提供了上述rna片段在检测细胞内hur转录后调控活性中的应用。
本发明还提供了一种用于检测细胞内hur转录后调控活性的方法,其包括以下步骤:
s1:将包含报告基因表达框的报告基因系统导入所述细胞,所述报告基因表达框的3’-utr区包含seqidno:1所示的dna片段;
s2:通过检测所述报告基因的表达来计算所述细胞内hur转录后调控活性。可将报告基因的表达强度作为指示hur转录后调控活性的指标。
优选地,所述报告基因系统为双荧光素酶报告基因系统,包括荧光素酶i表达框和荧光素酶ii表达框,所述dna片段连接在荧光素酶i表达框的3’-utr区,所述荧光素酶i激发产生的荧光波长与荧光素酶ii不同。将hur转录后调控活性表示为荧光素酶i激发产生的荧光强度与荧光素酶ii激发产生的荧光强度的比值。
优选地,所述荧光素酶i表达框和荧光素酶ii表达框处于同一个载体上。
优选地,所述双荧光素酶报告基因系统通过将所述dna片段插入至质粒psicheck中海肾荧光素酶基因的3’-utr区得到。
优选地,s1具体包括:
s11:将所述细胞培养至贴壁,并恢复形态;
s12:将所述报告基因系统转染至细胞中。
优选地,s2具体包括:
s21:转染后将细胞继续培养24-36小时,洗涤细胞;
s22:分别检测转染后的细胞中的荧光素酶i和荧光素酶ii的活性;
s23:将荧光素酶ii活性归一化荧光素酶i活性得到值作为衡量hur转录后调控活性的指标。
通过使用本发明的rna片段和方法,可直接针对性地检测细胞内hur的转录后调控活性,而非仅仅检测其转录本或蛋白质的含量,从而使得能够更准确地分析hur作为mrna结合因子在一些发育生物学和病理学发展中所起的作用。
附图说明
图1为xhoi与noti对重组质粒进行双酶切后的琼脂糖凝胶电泳照片;
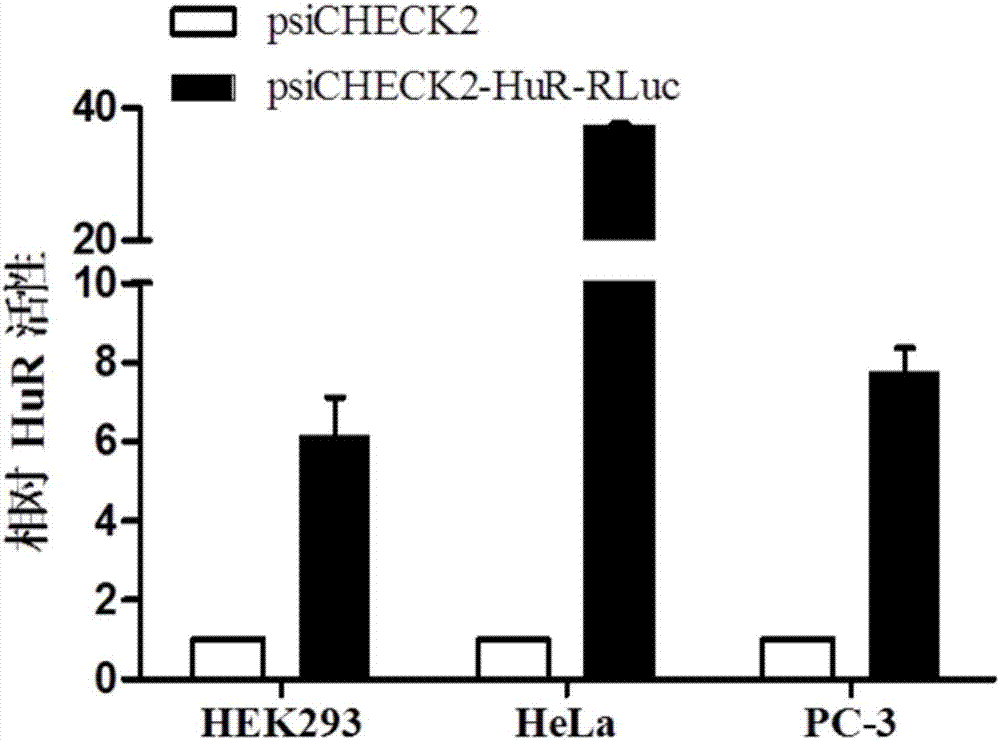
图2为分别转染有psicheck2和psicheck2-hur-rlu的人前列腺癌细胞系pc-3、人宫颈癌细胞系hela和人胚肾细胞系hek-293t细胞中的相对hur活性(即,萤火虫荧光素酶活性与海肾荧光素酶活性的比值)的统计图;
图3为分别转染有pcdna-tap(即,空载体)和pcdna-tap-hur的人前列腺癌细胞系pc-3、人宫颈癌细胞系hela和人胚肾细胞系hek-293t细胞中的相对hur活性(即,萤火虫荧光素酶活性与海肾荧光素酶活性的比值)的统计图。
具体实施方式
以下结合实例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
1.构建hur蛋白可结合的rna片段的dna编码序列
发明人对hur进行研究分析,找到hur结合的mrna核心序列,参考该序列设计出编码hur蛋白可结合的rna片段的dna编码序列,其序列如seqidno:1所示。将其连接至psichecktm-2vector海肾荧光素酶基因(hrluc)下游位点,即hrluc3'-utr区,组成hrluc-hurtargetrnamotif-hluc双荧光素酶报告基因质粒。
为保证载体构建的成功,在末端加入相应的限制性内切酶酶切位点的粘性末端,本例在5’端加入xhoi酶切位点的粘性末端(ctcgag),在3’端加入noti酶切位点的粘性末端(gcggccgc),序列两边的粘性末端的设计有助于片段与载体的连接。
通过从头合成的方法,分别合成该片段的两条寡核苷酸链,其序列分别如seqidno:2和3所示。两条寡核苷酸链互补配对后形成5’端包含xhoi酶切位点粘性末端和3’端包含noti酶切位点粘性末端的双链dna。100μl退火体系如下:annealingbufferfordnaoligos(5x)20μl、寡核苷酸链(50μm)各20μl,余下为ddh2o。
充分混匀后,设置pcr仪程序进行退火反应,具体程序为:95℃退火2分钟(目的是让dnaoligo充分变性);每90秒下降1℃,降至25℃后结束反应。dna退火产物用1.5%普通琼脂糖凝胶电泳鉴定,测浓度后置冰上备用。
2.将上述dna片段插入荧光素酶表达载体
本发明实施例选用的荧光素酶表达报告基因质粒为psichecktm2质粒。用限制性内切酶xhoi和noti(购自takara公司,xhoi货号为1635,noti货号为1623)对上述psichecktm2质粒进行双酶切,20μl酶切体系如下:10xquickcutgreenbuffer2μl,xhoi1μl,noti1μl,psichecktm2空载体1μg,余下为ddh2o。
充分混匀后,37℃酶切反应90分钟,用1.5%普通琼脂糖凝胶电泳鉴定质粒双酶切后的情况,然后切胶回收(dna胶回收试剂盒购自天根生化科技有限公司,货号为dp209),回收结束后测样品浓度,置冰上备用。
将退火得到的双链dna连接至psichecktm2质粒荧光素酶报告基因质粒上。10μl连接体系如下:dna连接酶(购自takara公司,货号为6022)5μl,双链dna片段与经双酶切的psichecktm2载体共5μl。为促进酶连成功率,将dna片段与载体的摩尔比控制在8:1左右。充分混匀后,将酶连体系置于pcr仪中,16℃反应1小时,连接反应结束后得到重组质粒,置于冰上备用。
将重组质粒(含有编码hur蛋白可结合位点的dna序列的psichecktm2报告基因质粒)转化至大肠杆菌。具体方法如下:取解冻后的感受态大肠杆菌dh5α,加入上述连接好的重组质粒,轻柔吹打,冰上孵育20分钟,42℃热休克60秒,冰上静置3分钟,然后加入450μl不含抗生素的lb液体培养基,放置于恒温摇床中复苏1小时(37℃,200rpm)。取复苏后的菌液至铺有固体培养基(含氨苄青霉素)培养皿上,用玻璃铺菌器将菌液均匀地铺满整个平皿,放置10分钟后,倒置于37℃孵箱过夜后,观察细菌生长情况。挑取菌落至5ml含氨苄青霉素的lb液体培养基,摇菌(37℃,200rpm)繁殖12小时后提取质粒,1.5%普通琼脂糖凝胶电泳鉴定。
用内切酶xhoi和noti对上述提取的质粒进行双酶切鉴定,本发明实施例双酶切鉴定如图1所示(泳道1为marker,泳道1-3显示为阳性克隆)。阳性组送至武汉擎科生物技术有限公司测序,确认dna片段已整合至psichecktm-2vector报告基因质粒上。至此,含有编码hur蛋白可结合位点的dna片段的psichecktm2报告基因质粒(以下均表示为psicheck2-hur-rluc)构建成功。
3.psicheck2-hur-rluc转染细胞
本发明实施例采用人前列腺癌细胞系pc-3、人宫颈癌细胞系hela、人胚肾细胞系hek-293t进行双荧光素酶实验。分别将对数期生长的细胞均匀种植在24孔板内,每孔约10万个细胞,用10%胎牛血清,高糖dmem培养基培养过夜。待细胞贴壁并恢复形态后,将报告基因系统相关质粒转染至细胞中(转染时细胞密度约为60-80%为宜)。使用的转染试剂为neofect(购自零客创智生物科技有限公司,货号为tf20121201)。50μl转染体系如下:质粒0.5μg,neofect转染试剂0.5μl,余下为无血清培养基。
本实验中使用的质粒分别为:psichecktm-2vector报告基因质粒、psicheck2-hur-rluc(含有编码hur蛋白可结合位点的dna片段的psichecktm-2vector报告基因质粒)、pcdna-tap(空质粒)、pcdna-tap-hur(hur过表达质粒)。
4.计算hur的活性
以萤火虫荧光素酶活性作为内参进行标准化,计算hur活性。具体实验方法如下:转染后的细胞继续培养24-36小时,再弃培养基上清,用1xpbs清洗细胞3次,每次5分钟,清洗细胞时注意操作轻柔,避免正面吹打细胞。采用双荧光素酶报告基因检测试剂盒(购自盖宁生物科技有新公司,货号为gn201-01)进行检测。
本发明实施例为验证该报告基因系统是否具有活性,将psichecktm-2质粒和psicheck2-hur-rluc质粒转染至细胞中,检测报告基因系统的活性,测定结果如图2所示,处理组(转染psicheck2-hur-rluc)的相对荧光素酶活性明显高于对照组(转染psichecktm-2空载体)。
本发明实施例为验证该报告基因系统是否可以特异性检测hur活性,将pcdna-tap质粒和pcdna-tap-hur质粒分别转染至细胞,结果如图3所示,外源导入hur后,阳性处理组(转染pcdna-tap-hur)荧光活性明显高于对照组(转染pcdna-tap空载体)。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
序列表
<110>华中科技大学同济医学院附属协和医院
<120>一种hur蛋白可识别的rna片段及在hur活性检测中的应用
<130>1
<160>3
<170>patentinversion3.5
<210>1
<211>66
<212>dna
<213>人工序列
<400>1
tggataccgttcgttgttaaccgttgatagccttggtttttaagccgtatatggctgtaa60
ataatt66
<210>2
<211>73
<212>dna
<213>人工序列
<400>2
tcgagtggataccgttcgttgttaaccgttgatagccttggtttttaagccgtatatggc60
tgtaaataattgc73
<210>3
<211>73
<212>dna
<213>人工序列
<400>3
ggccgcaattatttacagccatatacggcttaaaaaccaaggctatcaacggttaacaac60
gaacggtatccac73
- 还没有人留言评论。精彩留言会获得点赞!