抗AXL抗体、抗体片段和它们的免疫缀合物以及其用途的制作方法
本发明涉及抗axl抗体、抗体片段和此类抗体及抗体片段的免疫缀合物以及此类抗体、抗体片段和免疫缀合物在诊断和治疗方法中的用途。
背景技术:
axl蛋白(也称为ark、ufo、tyro-7)是tyro-3激酶家族中的受体酪氨酸激酶。tyro-3受体激酶的特征在于细胞外区域中两个免疫球蛋白样结构域和双纤连蛋白iii型重复与细胞质激酶结构域的组合。tyro-3受体激酶的配体是gas6(生长停滞特异性6)和蛋白s,它们是两种维生素k依赖性蛋白,显示出43%的氨基酸序列同一性并具有相似的结构域结构。每种蛋白都具有一个含有11g-羧基谷氨酸残基的n末端gia结构域,随后是四个表皮生长因子(egf)样模块,以及由两个串联层粘连蛋白g结构域组成的c末端性激素结合球蛋白(shbg)样结构。shbg结构域对于tyro-3受体激酶结合和活化而言是必要且充分的,而gia结构域结合带负电荷的膜磷脂并在tyro-3激酶介导的对凋亡细胞的吞噬中起重要作用。
axl活化导致通过pi-3-激酶/akt(franke等,oncogene,第22卷,第8983-8998页,2003)和其他诸如ras/erk和β-连环蛋白/tcf的主要途径的信号传导(goruppi等,mol.cell.biol,第21卷,第902-915页,2001)。axl在一系列正常组织(包括脑、心脏、骨骼肌、器官囊和几种其他器官的结缔组织)中,以及单核细胞中弱表达,但在淋巴细胞中不是如此。已经在成纤维细胞(goruppi等,mol.cell.biol.,第17卷,第4442-4453页,1997)、内皮细胞(hasanbasic等,amjphysiolheartcircphysiol,第287卷,h1207-h1213,2004)、血管平滑肌细胞(melaragno等,j.mol.cell.cardiol.,第37卷,第881-887页,2004)和神经元(allen等,mol.endocrinol.,第13卷,第191-201页,1999)的存活中描述了axl诱导的akt磷酸化。此外,axl在细胞粘附和趋化性中起作用,原因是axl基因敲除动物由于血小板整联蛋白iib3的活化减少而显示出受损的血小板聚集体稳定化作用和血栓形成。
axl或其配体gas6的失调涉及多种人类癌症的发病机理。axl过度表达已经在各种癌症类型中得到了证实,例如乳腺癌(meric等,clin.cancerres.,第8卷,第361-367页,2002;berclaz等,ann.oncol.,第12卷,第819-824页,2001)、结肠癌(chen等,int.j.cancer,第83卷,第579-584页,1999;craven等,int.j.cancer,第60卷,第791-797页,1995)、前列腺癌(jacob等,cancerdetect.prey.,第23卷,第325-332页,1999)、肺癌(wimmel等,eurjcancer,第37卷,第2264-2274页,2001)、胃癌(wu等,anticancerres.,第22卷,第1071-1078页,2002)、卵巢癌(sun等,oncology,第66卷,第450-457页,2004)、子宫内膜癌(sun等,ann.oncol.,第14卷,第898-906页,2003)、肾癌(chung等,dnacellbiol.,第22卷,第533-540页,2003)、肝细胞癌(tsou等,genomics,第50卷,第331-340页,1998)、甲状腺癌(ito等,thyroid,第12卷,第971-975页,2002;ito等,thyroid,第9卷,第563-567页,1999),此外,在慢性髓性白血病(janssen等,oncogene,第6卷,第2113-2120页,1991;braunger等,oncogene,第14卷,第2619-2631页,1997;o’bryan等,mol.cell.biol.,第11卷,第5016-5031页,1991)、急性髓性白血病(rochlitz等,leukemia,第13卷,第1352-1358页,1999)、骨肉瘤(nakano等,j.biol.chem.,第270卷,第5702-5705页,2003)、黑色素瘤(vanginkel等,cancerres.,第64卷,第128-134页,2004)、以及头颈部鳞状细胞癌(green等,brj.cancer.,第94卷,第1446-5页,2006)中得到了证实。
最近,通过分析磷酸酪氨酸信号传导,在约5%的nsclc原发性肿瘤中检测到活化的axl(rikova等,cell,第131卷,第1190-1203页,2007页)。靶向化疗药物诱导axl表达,药物诱导的axl表达使急性髓细胞白血病具有化疗耐药性(hong等,cancerletters,第268卷,第314-324页,2008),以及使胃肠道间质肿瘤(mehadevan等,oncogene,第26卷,第3909-3919页,2007)和乳腺癌(liu等,cancerresearch,第281卷,第6871-6878页,2009)分别具有对伊马替尼和拉帕替尼/赫赛汀的耐药性。
此外,已经确定axl与肿瘤转移相关,因为与非侵入性细胞相比,axl在侵袭性乳腺癌细胞系中上调。在体外,发现axl活性是迁移和侵袭所需要的,并且该活性可以通过抗体处理来抑制(wo04/008147)。类似地,在小鼠异种移植实验中,通过表达显性失活形式的axl(vajkoczy,p.等,proc.natl.acad.scienceu.s.a.,第103卷,第5799-5804页,2005)或者通过sirna介导的axl下调(holland等,cancerres.,第65卷,第9294-9303页,2005)体内消除axl活性,防止了皮下和原位细胞生长。
因此,已经描述了抗axl单克隆抗体用于治疗癌症。例如,涉及抗axl抗体的公开包括wo2009/063965、wo2009/062690、wo2011/014457、us2014/0227283和第8,853,369号美国专利。us2014/0227283公开了抗axl单克隆抗体及其在诊断和治疗方法中的用途。wo2009/062690公开了结合axl蛋白的细胞外结构域并且可以至少部分抑制axl活性的抗体。
这些抗axl单克隆抗体将在患者身体的任何位置以相似的亲和力结合ax1,包括它们意图治疗的肿瘤的位置。预期这些抗体在非肿瘤环境中与ax1的结合会对这些环境中axl的正常功能产生不利影响,因此可能会引起显著的副作用。本发明提供条件活性的抗axl抗体和抗体片段,与其在非肿瘤环境中对axl的结合亲和力相比,其在肿瘤微环境中对ax1具有更高的结合亲和力。与本领域已知的抗axl单克隆抗体相比,预期本发明的抗axl抗体和抗体片段具有相当的或更高的抗癌功效并且具有减少的副作用。这还可以允许施用更高剂量的抗axl抗体和抗体片段或更频繁的治疗,从而提供更有效的治疗选择。
技术实现要素:
在一个方面,本发明提供了分离的重链可变区多肽,其特异性结合axl蛋白。该多肽包括三个互补决定区h1、h2和h3序列,其中:
h1序列为x1gx2x3mx4(seqidno:1);
h2序列为likx5snggtx6ynqkfkg(seqidno:2);
h3序列为gx7x8x9x10x11x12x13x14dyx15x16(seqidno:3),
其中,
x1为t或a或w,
x2为h或a,
x3为t或i,
x4为n或i,
x5为p或n,
x6为s或i或t,
x7为h或d或e或p或r或w,
x8为y或n,
x9为e或a或d或f或g或h或i或l或m或n或r或v或y,
x10为s或d或m或n或q,
x11为y或c或e或p,
x12为f或e或n或s或t或v,
x13为a或d或g或l或y,
x14为m或e或f,
x15为w或a或d或h或l或n或p或r或t,
x16为g或h。
在另一个方面,分离的重链可变区多肽与分离的轻链可变区组合,所述分离的轻链可变区包括三个互补决定区l1、l2和l3序列,其中:
l1序列为kasqdx17x18sx19vx20(seqidno:4);
l2序列为x21x22x23trx24t(seqidno:5);
l3序列为qex25x26sx27x28x29x30(seqidno:6),
其中,
x17为v或d或g或n或w,
x18为s或v,
x19为a或l或m,
x20为a或d或n或q,
x21为w或f,
x22为a或i或n或p或q,
x23为s或d,
x24为h或d,
x25为h或c或f或i或l或q或s或t或v或y,
x26为f或c或d或e或g或n或s,
x27为t或c或p,
x28为p或a或c或d或e或h或k或s或t或v或w,
x29为l或g或r,
x30为t或i或r。
在另一方面,本发明提供了抗axl抗体或抗体片段,其包括本发明的分离的重链可变区多肽。
在另一方面,本发明提供免疫缀合物,其包括本发明的抗体或抗体片段,其任选地与选自化学治疗剂、放射性原子、细胞抑制剂和细胞毒性剂的药剂缀合。
在另一方面,本发明提供药物组合物,其包括本发明的多肽、抗体或抗体片段或免疫缀合物,以及药学上可接受的载体。
在另一方面,本发明提供了用于诊断或治疗的试剂盒,其包括本发明的多肽、抗体或抗体片段或免疫缀合物。
附图说明
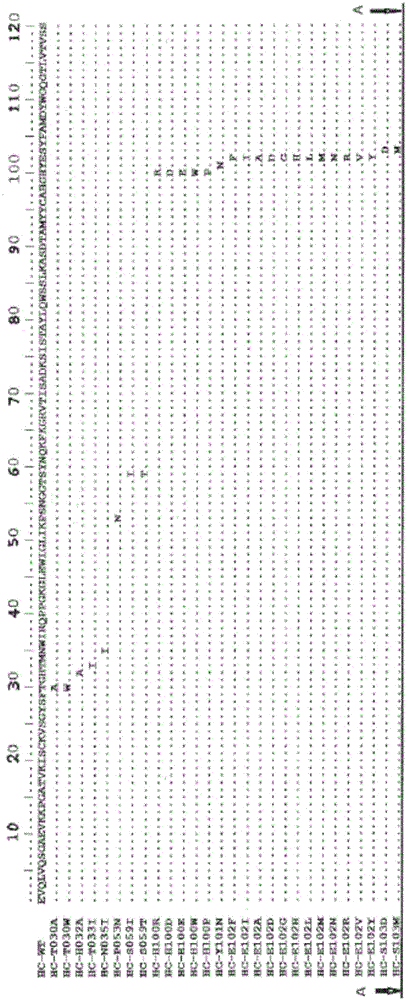
图1a-1b分别显示了本发明的抗axl抗体的重链可变区和轻链可变区的序列比对。
图2显示了在ph6.0和ph7.4下本发明的各种条件活性抗体与axl的细胞外结构域的结合(od450)。这些条件活性抗体在ph6.0下比在ph7.4下更具活性。
图3显示了本发明的各种条件活性抗体对axl的细胞外结构域的选择性。选择性测定为在ph6.0下对结合配偶体的结合亲和力与在ph7.4下对相同结合配偶体的结合亲和力的比。
图4显示了通过尺寸排阻色谱表明本发明的条件活性抗体不聚集,如实施例1中所述。
图5显示了通过elisa测定法测量的热休克之前和热休克之后的本发明的条件活性抗体的热稳定性,如实施例1中所述。
图6a-6b显示了实施例1中通过spr测定法测量的本发明的条件活性抗体的选择性。
图7显示了在krebs缓冲液中本发明的抗axl抗体与ax1结合的ph依赖性结合谱。
图8a-8e显示了使用a549细胞的另一种细胞杀伤研究的结果,其中本发明的抗axl抗体用于在ph6.0和ph7.4下以不同的抗体浓度进行细胞杀伤。
图9a-9d显示了本发明的抗axl抗体在不同缓冲液和不同ph水平下对人axl和食蟹猴axl的结合亲和力。
图10a-10h显示了与金霉素(duomycin)缀合的本发明的抗axl抗体在不同ph水平下对不同细胞系的细胞杀伤。
图11显示了与吉西他滨缀合的本发明的抗axl抗体在不同ph水平下对a549细胞的细胞杀伤。
图12显示了使用金霉素缀合的本发明的抗axl抗体治疗异种移植小鼠,对肿瘤体积的影响。
图13a-13b显示了在注射缀合物后食蟹猴血液中检测到的金霉素缀合的本发明的抗axl抗体的存在随时间的变化。
图14a显示了从缀合物注射前(前(d-3))开始直至注射后3天(后(d-3)),食蟹猴血液中检测到的天冬氨酸转氨酶(ast)的存在随时间的变化。
图14b显示了从缀合物注射前(前(d-3))开始直至注射后3天(后(d-3)),食蟹猴血液中检测到的丙氨酸转氨酶(alt)的存在随时间的变化。
图15显示了注射缀合物后食蟹猴血液中淋巴细胞计数随时间的变化。
图16a-16b分别显示了接受lclc103h和du145的小鼠的体内治疗。
定义
为了便于理解本文提供的实施例,本文定义了某些频繁出现的术语。
本文中使用的与测定量有关的术语“约”是指测定量的正常变化,其可由进行测量并且其小心程度与测量目的和测量仪器精度挂钩的熟练人员预测。除非另有说明,“约”是指所提供值的变化为+/-10%。
本文中使用的术语“亲和力”是指分子(例如抗体)的单个结合位点与其结合配偶体(例如抗原)之间的非共价相互作用的总和的强度。除非另有说明,否则如本文中使用的,“结合亲和力”是指固有的(intrinsic)结合亲和力,其反映结合对的成员(例如,抗体和抗原)之间的1:1相互作用。分子x对其配偶体y的亲和力通常可由解离常数(kd)表示。亲和力可以通过本领域已知的常规方法测量,包括本文所述的那些方法。用于测量结合亲和力的具体说明性和示例性实施方案描述如下。
在提及抗体时使用的时候,术语“亲和力成熟”是指在一个或多个高变区(hvr)中具有一个或多个改变的抗体或抗体片段,与不具有这种改变的亲本抗体或抗体片段相比,这种改变导致抗体或抗体片段对抗原的亲和力提高。
本文中使用的术语“氨基酸”是指任何含有氨基(-nh2)和羧基(-cooh)的有机化合物;氨基和羧基优选为游离基或缩合后作为肽键的一部分。本领域已知的“二十个自然编码的、形成多肽的、α-氨基酸”是指:丙氨酸(ala或a)、精氨酸(arg或r)、天冬酰胺(asn或n)、天冬氨酸(asp或d)、半胱氨酸(cys或c)、谷氨酸(glu或e)、谷氨酰胺(gln或q)、甘氨酸(gly或g)、组氨酸(his或h)、异亮氨酸(ile或i)、亮氨酸(leu或l)、赖氨酸(lys或k)、蛋氨酸(met或m)、苯丙氨酸(phe或f)、脯氨酸(pro或p)、丝氨酸(ser或s)、苏氨酸(thr或t)、色氨酸(trp或w)、酪氨酸(tyr或y)和缬氨酸(val或v)。
本文中使用的术语“抗血管生成剂”是指在一定程度上阻断或干扰血管形成的化合物。例如,抗血管生成剂可以是与参与促进血管生成的生长因子或生长因子受体结合的小分子或抗体。在一个实施方案中,抗血管生成剂是与血管内皮生长因子(vegf)结合的抗体或抗体片段,例如贝伐单抗
本文中使用的术语“抗体片段”是指除完整抗体以外的分子,其包含完整抗体的一部分,该部分结合与完整抗体结合的抗原。抗体片段的实例包括但不限于fv、fab、fab’、fab’-sh、f(ab’)2;双体抗体(diabodies);线性抗体;单链抗体分子(例如scfv)。这些抗体片段保留了一些选择性结合作为其来源的抗体的抗原(例如,多肽抗原)的能力,可以使用本领域熟知的方法制备(参见,例如,harlow和lane,同上)。
本文中使用的术语“抗体”是指完整的免疫球蛋白分子。抗体或抗体片段可用于通过免疫亲和层析分离制备量的抗原。这种抗体或抗体片段的各种其他用途为诊断疾病和/或对疾病分期(例如瘤形成)和用于治疗疾病的治疗应用,所述疾病例如:瘤形成、自身免疫疾病、aids、心血管疾病、感染等。嵌合抗体、类人抗体、人源化抗体或全人源抗体或抗体片段特别有利于施用给人类患者。
fab片段由抗体分子的一价抗原结合片段组成,并且可以通过用木瓜蛋白酶消化完整抗体分子以产生由完整轻链和部分重链组成的片段来制备。
抗体分子的fab’片段可以通过用胃蛋白酶处理整个抗体分子,然后还原得到由完整轻链和部分重链组成的分子而获得。每个以这种方式处理的抗体分子获得两个fab’片段。
抗体的(fab’)2片段可以通过用胃蛋白酶处理整个抗体分子但不进行随后的还原而获得。(fab’)2片段是通过两个二硫键保持在一起的两个fab’片段的二聚体。
fv片段定义为含有表达为两条链的轻链可变区和重链可变区的基因工程片段。
本文中使用的术语“抗axl抗体”,抗axl抗体片段和“结合axl的抗体或抗体片段”是指能够以足够的亲和力结合axl的抗体或抗体片段,使得抗体或抗体片段可用作靶向axl的诊断剂和/或治疗剂。在一个实施方案中,抗axl抗体或抗体片段与不相关的非axl蛋白的结合程度小于(例如通过放射免疫测定法ria)测得的该抗体或抗体片段与ax1的结合程度的约10%。在某些实施方案中,结合axl的抗体或抗体片段具有≤1μm、≤100nm、≤10nm、≤1nm、≤0.1nm、≤0.01nm或≤0.001nm的解离常数(kd)(例如10-8m或更低,或10-8m至10-13m,或10-9m至10-13m)。在某些实施方案中,抗axl抗体或抗体片段结合axl的表位,该表位在来自不同物种的axl中是保守的。
本文中使用的术语“血管生成疾病”是指血管生成的任何失调,包括非肿瘤和肿瘤病症。肿瘤病症包括但不限于下面描述的那些(参见例如“癌症”)。非肿瘤性疾病包括但不限于不希望的或异常的肥大、关节炎、类风湿性关节炎(ra)、牛皮癣、银屑病斑块、结节病、动脉粥样硬化、动脉粥样硬化斑块、糖尿病、其他增生性视网膜病,包括早产儿视网膜病、晶状体后纤维组织增生症、新生血管性青光眼、年龄相关性黄斑变性、糖尿病性黄斑水肿、角膜新生血管、角膜移植新生血管、角膜移植排斥、视网膜/脉络膜新生血管、角部新生血管形成(虹膜发红)、眼新生血管疾病、血管再狭窄、动静脉畸形(avm)、脑膜瘤、血管瘤、血管纤维瘤、甲状腺增生(包括格雷夫斯病)、角膜及其他组织移植、慢性炎症、肺部炎症、急性肺损伤/ards、败血症、原发性肺动脉高压、恶性肺积液、脑水肿(例如与急性脑卒中/闭合性头部受伤/创伤有关)、滑膜炎症、类风湿性关节炎的血管翳形成、骨化性肌炎、骨质增生性骨形成、骨关节炎(oa)、难治性腹水、多囊卵巢疾病、子宫内膜异位症、第三间隙液病(3rdspacingoffluiddisease)(胰腺炎、室间隔综合征、烧伤、肠道疾病)、子宫肌瘤、早产、慢性炎症例如ibd(克罗恩病和溃疡性结肠炎)、肾同种异体移植物排斥、炎症性肠病、肾病综合征、不希望的或异常的组织肿块生长(非癌症)、血友病性关节、肥厚性瘢痕、毛发生长抑制、奥韦综合征(osler-weber综合征)、化脓性肉芽肿、晶状体后纤维增生、硬皮病、沙眼、血管粘连、滑膜炎、皮炎、先兆子痫、腹水、心包积液(例如与心包炎相关)和胸腔积液。
本文中使用的术语“血管生成”是指有助于从预先存在的血管生长新血管(特别是但不限于提供新的供应肿瘤的血管)的所有涉及axl的过程。这些过程包括多种细胞事件,例如血管内皮细胞的增殖、存活、迁移和出芽,周细胞的吸引和迁移,以及用于血管稳定、血管灌注或分泌血管生成因子的基底膜形成(由基质细胞或肿瘤细胞形成),并且这些过程应通过axl的非催化活性或催化活性刺激或介导,优选包括axl磷酸化和/或axl介导的信号转导。
除非另有说明,本文中使用的术语“axl”是指来自任何脊椎动物来源的任何天然ax1,包括哺乳动物例如灵长类动物(例如人)和啮齿动物(例如小鼠和大鼠)。该术语包括“全长”未加工的axl以及由细胞中的加工产生的任何形式的axl。该术语还包括axl的天然存在的变体,例如剪接变体或等位基因变体。人axl的氨基酸序列在本领域中是公知的,可从例如genbank的公共数据库获得。
本文中使用的术语“axl活化”是指axl受体的活化或磷酸化。通常,axl活化导致信号转导(例如由axl或底物多肽中的axl受体磷酸化酪氨酸残基的细胞内激酶结构域引起的信号转导)。axl活化可以通过axl配体(gas6)与目的axl受体结合来介导。与axl结合的gas6可以激活axl的激酶结构域,从而导致axl中酪氨酸残基的磷酸化和/或其他底物多肽中酪氨酸残基的磷酸化。
本文中使用的术语“axl介导的抗细胞凋亡”是指防止人类细胞(优选但不限于人类癌细胞)的程序性细胞死亡(细胞凋亡)的所有涉及axl的过程。特别地,它指的是这样的过程,该过程防止人类细胞(优选但不限于人类癌细胞)通过生长因子戒断、缺氧、暴露于化学治疗剂或辐射,或者启动fas/apo-1受体介导的信号传导而诱导细胞凋亡,并且该过程通过axl的非催化活性或催化活性刺激或介导,优选包括axl磷酸化和/或axl介导的信号传导。
本文中使用的术语“结合”是指抗体的可变区或fv与抗原的相互作用,其依赖于抗原上的特定结构(例如,抗原决定簇或表位)的存在。例如,抗体可变区或fv通常识别并结合特定蛋白结构而不是蛋白。本文中使用的术语“特异性结合”或“特异地结合”是指与其他蛋白相比,抗体可变区或fv以更频繁、更快速的方式以更长的持续时间和/或更大的亲和力与特定抗原结合或连接。例如,相比于抗体可变区或fv与其他抗原的结合,该可变区或fv与其抗原的特异性结合亲和力、抗体亲抗原性更大,结合更容易,和/或持续时间更长。再例如,抗体可变区或fv与细胞表面蛋白(抗原)结合的亲和力实质上大于其对相关蛋白或其他细胞表面蛋白或对多反应性天然抗体(即天然存在的、已知可以结合人体中天然存在的各种抗原的抗体)通常识别的抗原的亲和力。然而,“特异性结合”不一定需要排他性结合或不可检测到的对另一种抗原的结合,这是术语“选择性结合”的含义。在一个实例中,抗体可变区或fv(或其他结合区)与抗原的“特异性结合”,意指抗体可变区或fv与抗原结合的平衡常数(kd)为100nm或更小,例如50nm或更小,例如20nm或更小,例如15nm或更小,或10nm或更小,或5nm或更小,2nm或更小,或1nm或更小。
本文中使用的术语“癌症”和“癌性”是指或者描述的是哺乳动物中通常以不受调节的细胞生长/增殖为特征的生理状况。癌症的实例包括但不限于癌、淋巴瘤(例如,霍奇金氏淋巴瘤和非霍奇金氏淋巴瘤)、胚细胞瘤、肉瘤和白血病。这类癌症的更具体的例子包括鳞状细胞癌、小细胞肺癌、非小细胞肺癌、肺腺癌、肺鳞状细胞癌、腹膜癌、肝细胞癌、胃肠癌、胰腺癌、神经胶质瘤、宫颈癌、卵巢癌、肝癌、膀胱癌、肝细胞瘤、乳腺癌、结肠癌、结直肠癌、子宫内膜癌或子宫癌、唾液腺癌、肾癌、肝癌、前列腺癌、外阴癌、甲状腺癌、肝肿瘤、白血病、其他淋巴组织增生性疾病、以及各种类型的头颈癌。
本文中使用的术语“细胞增生性疾病”和“增生性疾病”是指与某种程度的异常细胞增生相关的疾病。在一个实施方案中,细胞增生性疾病是癌症。
本文中使用的术语“化学治疗剂”是指可用于治疗癌症的化学化合物。化学治疗剂的实例包括烷化剂,例如噻替哌和环磷酰胺
本文定义的化学治疗剂包括“抗激素剂”或“内分泌治疗剂”,其用于调节、减少、阻断或抑制可促进癌症生长的激素的作用。它们本身可以是激素,包括但不限于:具有混合激动剂/拮抗剂谱的抗雌激素,包括他莫昔芬
本文中使用的术语“嵌合”抗体是指其中的重链和/或轻链的一部分来源于特定的来源或物种,而重链和/或轻链的其余部分来源于不同的来源或物种的抗体。
本文中使用的术语抗体“类别”是指其重链所具有的恒定结构域或恒定区的类型。有五种主要类别的抗体:iga、igd、ige、igg和igm,其中的几种可以进一步分为亚类(同种型),例如igg1、igg2、igg3、igg4、iga1和iga2。对应于免疫球蛋白的不同类别的重链恒定区分别称为α、δ、ε、γ和μ。
本文中使用的术语“条件活性抗体”和“条件活性抗体片段”是指在肿瘤微环境中的一种条件的一个值下,与在非肿瘤微环境中相同条件的不同值下相比,更具活性的抗体或抗体片段。与非肿瘤微环境中的条件相比,肿瘤微环境中的条件可包括较低的ph、较高浓度的乳酸和/或丙酮酸、低氧、较低浓度的葡萄糖和稍高的温度。例如,在一个实施方案中,条件活性抗体或抗体片段在正常体温下实质上可以是无活性的,但在肿瘤微环境中可遇到的较高温度下具有活性。在另一个实施方案中,条件活性抗体或抗体片段在正常含氧血液中的活性可低于可存在于肿瘤微环境中的低氧环境中的活性。在本领域技术人员已知的肿瘤微环境中还存在其他条件,它们也可以被选择用作本发明中的条件,其可以触发抗axl抗体或抗体片段在该条件的不同值下具有不同的活性。
本文中使用的术语“组成型”,例如当用于axl活性时,是指受体激酶的连续的信号传导活性,其不依赖于配体或其他活化分子的存在。取决于受体激酶的性质,所有活性都可以是组成型的,或者受体的活性可以通过其他分子(例如配体)的结合进一步活化。导致受体激酶活化的细胞事件是本领域普通技术人员所熟知的。例如,活化可以包括寡聚化,例如二聚化、三聚化等,以形成更高级的受体复合物。复合物可包含单一种类的蛋白,即同质型复合物。或者,复合物可包含至少两种不同的蛋白,即异质型复合物。复合物形成可以由例如细胞表面上正常或突变形式的受体的过表达引起。复合物形成也可能由受体中的特定突变或突变引起。
本文中使用的术语“细胞抑制剂”是指在体外或体内阻止细胞生长的化合物或组合物。因此,细胞抑制剂可以是显著降低s期细胞百分比的细胞抑制剂。细胞抑制剂的其他实例包括通过诱导g0/g1停滞或m期停滞来阻断细胞周期进展的制剂。人源化抗her2抗体曲妥珠单抗
本文中使用的术语“细胞毒性剂”是指抑制或阻止细胞功能和/或引起细胞死亡或损伤的物质。细胞毒性剂包括但不限于放射性同位素(例如,at211、i131、i125、y90、re186、re188、sm153、bi212、p32、pb212和lu的放射性同位素);化学治疗剂或药物(例如甲氨蝶呤、阿霉素、长春花生物碱(长春新碱、长春花碱、依托泊苷)、多柔比星、美法仑、丝裂霉素c、苯丁酸氮芥、柔红霉素或其他嵌入剂);生长抑制剂;酶及其片段例如核溶酶;抗生素;毒素例如小分子毒素或细菌、真菌、植物或动物来源的酶活性毒素,包括其片段和/或变体;以及下面公开的各种抗肿瘤剂或抗癌剂。
本文中使用的术语“双体抗体”是指具有两个抗原结合位点的小抗体片段,该片段包含与同一多肽链(vh-vl)中的轻链可变结构域(vl)连接的重链可变结构域(vh)。通过使用一种由于太短而不允许同一链上的两个可变结构域之间配对的接头,所述结构域被迫与另一条链的互补结构域配对并产生两个抗原结合位点。
本文中使用的术语“可检测标记”是指通过物理或化学方式直接或间接检测或测量的任何物质,其指示样品中ctc的存在。有用的可检测标记的代表性实例包括但不限于以下:基于光吸收、萦光、反射、光散射、磷光或发光性质可直接或间接检测的分子或离子;可通过其放射性质检测到的分子或离子;通过其核磁共振或顺磁性质可检测的分子或离子。例如,基于光吸收或萦光性可间接检测的分子的组中包括各种引起适当的底物转化的,例如从非光吸收分子转化为光吸收分子,或从非萦光分子转变为萦光分子。
本文中使用的术语“诊断”是指确定个体对疾病或病症的易感性,确定个体目前是否受疾病或病症影响,受疾病或病症影响的个体的预后(例如,鉴定转移前或转移性癌症状态、癌症阶段、或癌症对治疗的响应性)和度量疗法(例如,监测个体的状况以提供关于治疗的效果或功效的信息)。在一些实施方案中,本发明的诊断方法特别适用于检测早期癌症。
本文中使用的术语“诊断剂”是指可以直接或间接检测并用于诊断目的的分子。可以将诊断剂施用于个体或样本。诊断剂可以其本身提供,或者可以与载体例如条件活性抗体缀合。
本文中使用的术语“效应子功能”是指可归因于抗体fc区的那些生物活性,其随抗体同种型的不同而变化。抗体效应子功能的实例包括:c1q结合和补体依赖性细胞毒性(cdc);fc受体结合;抗体依赖性细胞介导的细胞毒性(adcc);吞噬作用;下调细胞表面受体(例如b细胞受体);和b细胞激活。
本文中使用的术语“有效量”,例如药物制剂的有效量,是指在必要的剂量和时间段内有效实现所需治疗结果或预防结果的量。
本文中使用的术语“fc区”用于定义免疫球蛋白重链的c末端区域,该c末端区域含有至少一部分恒定区。该术语包括天然序列fc区和变体fc区。在一个实施方案中,人igg重链fc区从cys226或从pro230延伸至重链的羧基末端。然而,fc区的c-末端赖氨酸(lys447)可以存在或不存在。除非本文另有说明,否则fc区或恒定区中氨基酸残基的编号依据eu编号系统,也称为eu索引,如kabat等,sequencesofproteinsofimmunologicalinterest,第5版,publichealthservice,nationalinstitutesofhealth,bethesda,md.,1991中所描述的。
本文中使用的术语“框架”或“fr”是指除高变区(重链中的hvr或h1-3和轻链中的l1-3)残基之外的可变结构域残基。可变结构域的fr通常由四个fr结构域组成:fr1、fr2、fr3和fr4。因此,hvr和fr序列通常以下列顺序出现在vh(或vl)中:fr1-h1(l1)-fr2-h2(l2)-fr3-h3(l3)-fr4。
术语“全长抗体”、“完整抗体”或“整个抗体”是指包含抗原结合可变区(vh或vl)以及轻链恒定结构域(cl)和重链恒定结构域ch1、ch2和ch3的抗体。恒定结构域可以是天然序列恒定结构域(例如人天然序列恒定结构域)或其氨基酸序列变体。根据全长抗体重链的恒定结构域的氨基酸序列,可以将全长抗体归为不同的“类别”。全长抗体有五种主要类型:iga、igd、ige、igg和igm,其中几种可进一步分为“亚类”(同种型),例如igg1、igg2、igg3、igg4、iga和iga2。对应于不同类别抗体的重链恒定结构域分别称为α、δ、ε、γ和μ。不同类别的免疫球蛋白的亚基结构和三维构型是众所周知的。
本文中使用的术语“宿主细胞”、“宿主细胞系”和“宿主细胞培养物”可互换使用,是指已引入外源核酸的细胞,包括此类细胞的后代。宿主细胞包括“转化体”和“转化细胞”,其包括原代转化细胞和由其衍生的后代,而不考虑传代次数。后代可能与亲本细胞的核酸含量不完全相同,但可含有突变。本文包括具有与在原代转化细胞中要筛选或选择的相同功能或生物活性的突变后代。
本文中使用的术语“人抗体”是具有如下氨基酸序列的抗体,所述氨基酸序列对应于人或人细胞产生的抗体的氨基酸序列,或者源自利用人抗体库或其它人抗体编码序列的非人来源的氨基酸序列。该人抗体的定义明确地排除了包含非人的抗原结合残基的人源化抗体。
本文中使用的术语“人共有框架”是代表在人免疫球蛋白vl或vh框架序列的选择对象中最常出现的氨基酸残基的框架。通常,人免疫球蛋白vl或vh序列的选择对象来自可变结构域序列的亚组。通常,序列亚组是如下出版物中所述的亚组:kabat等,sequencesofproteinsofimmunologicalinterest,第5版,nihpublication91-3242,bethesdamd.(1991),第1-3卷。在一个实施方案中,对于vl,亚组是如kabat等(同上)所述的亚组kappai。在一个实施方案中,对于vh,亚组是如kabat等(同上)所述的亚组iii。
本文中使用的术语“人源化”抗体是指嵌合抗体,其包含来自非人hvr的氨基酸残基和来自人fr的氨基酸残基。在某些实施方案中,人源化抗体包含基本上全部的至少一个可变结构域、通常两个可变结构域,其中全部或基本上全部的hvr(例如,cdr)对应于非人抗体的hvr,全部或基本上全部的fr对应于人抗体的fr。人源化抗体任选地可以包含衍生自人抗体的抗体恒定区的至少一部分。抗体,例如非人抗体,的“人源化形式”,是指已经历过人源化的抗体。
本文中使用的术语“高变区”或“hvr”是指抗体可变区中在序列上是高变的和/或形成结构上限定的环(“高变环”)的每个区域。通常,天然四链抗体包含六个hvr;vh中的三个(h1、h2、h3)和vl中的三个(l1、l2、l3)。hvr通常包含来自高变环和/或来自“互补决定区”(cdr)的氨基酸残基,后者具有最高的序列可变性和/或参与抗原识别。示例性高变环出现在氨基酸残基26-32(l1)、50-52(l2)、91-96(l3)、26-32(h1)、53-55(h2)和96-101(h3)(chothia和lesk,j.mol.biol.,第196卷,第901-917页,1987)。示例性cdr(cdr-l1,cdr-l2,cdr-l3,cdr-h1,cdr-h2和cdr-h3)出现在l1的24-34,l2的50-56,l3的89-97,h1的31-35b,h2的50-65和h3的95-102位氨基酸残基(kabat等,sequencesofproteinsofimmunologicalinterest,第5版,publichealthservice,nationalinstitutesofhealth,bethesda,md.1991)。除vh中的cdr1外,cdr通常包含形成高变环的氨基酸残基。cdr还包含为与抗原接触的残基的“特异性决定残基”或“sdr”。sdr包含在cdr中被称为缩略cdr(abbreviated-cdr)或a-cdr的区域内。示例性的a-cdr(a-cdr-l1、a-cdr-l2、a-cdr-l3、a-cdr-h1、a-cdr-h2和a-cdr-h3)出现在l1的31-34,l2的50-55,l3的89-96,h1的31-35b,h2的50-58,h3的95-102位氨基酸残基处(参见almagro和fransson,front.biosci.,第13卷,第1619-1633页,2008)。除非另有说明,否则可变结构域中的hvr残基和其他残基(例如,fr残基)在本文中根据kabat等(同上)编号。
本文中使用的术语“免疫缀合物”是与一个或多个异源分子缀合的抗体,包括但不限于细胞毒性剂。
本文中使用的术语“个体”或“受试者”是哺乳动物。哺乳动物包括但不限于驯养的动物(例如,牛、羊、猫、狗和马),灵长类动物(例如,人和非人灵长类动物,例如猴子)、兔子和啮齿动物(例如,小鼠和大鼠)。在某些实施方案中,个体或受试者是人。
本文中使用的术语“抑制细胞生长或增殖”是指将细胞的生长或增殖降低至少10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或100%,包括诱导细胞死亡。
本文中使用的术语“分离的”抗体是与其天然环境的组分分离的抗体。在某些实施方案中,将抗体纯化至大于95%或99%的纯度,所述纯度通过例如电泳(例如,sds-page、等电聚焦(ief)、毛细管电泳)或色谱法(例如,离子交换或反相hplc)测定。关于评估抗体纯度的方法的综述,参见,例如,flatman等,j.chromatogr.b,第848卷,第79-87页,2007。
本文中使用的术语“分离的”核酸是指已经与其天然环境的组分分离的核酸分子。分离的核酸包括通常含有核酸分子的细胞中包含的核酸分子,但该核酸分子存在于染色体外或存在于与其天然染色体位置不同的染色体位置上。
本文中使用的术语“编码抗axl抗体的分离的核酸”是指编码抗体重链和轻链(或其片段)的一个或多个核酸分子,包括在单一载体中或在各自载体中的核酸分子,以及存在于宿主细胞中一个或多个位置的此类核酸分子。
本文中使用的术语“配体非依赖性”,例如当用于受体信号传导活性时,是指不依赖于配体存在的信号传导活性。具有配体非依赖性激酶活性的受体未必排除配体与该受体的结合,从而产生激酶活性的额外活化。
本文中使用的术语“转移”是指支持癌细胞从原发性肿瘤扩散,渗透到淋巴和/或血管中,在血流中循环,并在身体其他部位的正常组织中的远处病灶中生长(转移)的所有涉及axl的过程。特别地,它指为转移的基础并且由axl的非催化活性或催化活性(优选包括axl磷酸化和/或axl介导的信号传导)刺激或介导的肿瘤细胞的细胞事件,例如增殖、迁移、非贴壁依赖性、细胞凋亡逃逸或血管生成因子的分泌。
本文中使用的术语“微环境”是指组织或身体的任何部分或区域,该部分或区域与组织的其他区域或身体的区域相比具有恒定的或暂时的物理或化学差异。对于肿瘤,本文中使用的术语“肿瘤微环境”是指肿瘤存在其中的环境,该环境是肿瘤内的非细胞区域和直接在肿瘤组织外侧但不属于癌细胞本身的细胞内区室的区域。肿瘤与肿瘤微环境密切相关,不断相互作用。肿瘤可以改变其微环境,微环境可以影响肿瘤的生长和扩散。通常,肿瘤微环境具有5.8至7.0范围内的低ph,更通常在6.2至6.8的范围内,最通常在6.4-6.8的范围内。另一方面,正常的生理ph通常在7.2-7.8的范围内。还已知与血浆相比,肿瘤微环境具有较低浓度的葡萄糖和其他营养物,但具有较高浓度的乳酸。此外,肿瘤微环境的温度可以比正常生理温度高0.3至1℃。肿瘤微环境已经描述于gillies等的“mriofthetumormicroenvironment”,journalofmagneticresonanceimaging,第16卷,第430-450页,2002。术语“非肿瘤微环境”是指除肿瘤以外的部位的微环境。
本文中使用的术语“单克隆抗体”是指从基本上同质的抗体的群体获得的抗体,即,除了例如含有天然存在的突变或者在单克隆抗体制剂的生产过程中产生的可能的抗体变体之外(这种变体通常以少量存在),包含该群体的各个抗体是相同的和/或结合相同的表位。与通常包括针对不同决定簇(表位)的不同抗体的多克隆抗体制剂相反,单克隆抗体制剂的每个单克隆抗体针对抗原上的单一决定簇。因此,修饰语“单克隆”表示抗体的特征是从基本上同质的抗体的群体获得的,并且不应解释为需要通过任何特定的方法产生抗体。例如,根据本发明使用的单克隆抗体可以通过多种技术制备,包括但不限于杂交瘤方法、重组dna方法、噬菌体展示方法、和利用含有全部或部分人免疫球蛋白基因座的转基因动物的方法,本文描述了此类方法和用于制备单克隆抗体的其他示例性方法。
本文中使用的术语“裸抗体(nakedantibody)”是指未与异源部分(例如,细胞毒性部分)或放射性标记缀合的抗体。裸抗体可以存在于药物制剂中。
本文中使用的术语“天然抗体”是指具有不同结构的天然存在的免疫球蛋白分子。例如,天然igg抗体是约150000道尔顿的异四聚体糖蛋白,由被二硫键连接的两条相同的轻链和两条相同的重链组成。从n-末端到c-末端,每条重链具有可变区(vh),也称为可变重链结构域或重链可变结构域,接着是三个恒定结构域(ch1、ch2和ch3)。类似地,从n-末端到c-末端,每条轻链具有可变区(vl),也称为可变轻链结构域或轻链可变结构域,然后是轻链恒定(cl)结构域。基于抗体轻链恒定结构域的氨基酸序列,抗体的轻链可以被归为两种类型中的一种,称为kappa(κ)和lambda(λ)。
本文中使用的术语“包装说明书”用于指通常包括在治疗产品的商业包装中的说明书,其包含关于适应症、用法、剂量、施用、联合治疗、禁忌症和/或关于使用这种治疗产品的警告的信息。
本文中使用的术语相对于参考多肽序列的“氨基酸序列同一性百分比(%)”定义为,在使序列对齐并在必要时引入空位以达成最大序列同一性百分比(不将任何保守性取代视为序列同一性的一部分)后,候选序列中与参考多肽序列中的氨基酸残基相同的氨基酸残基的百分比。用于确定氨基酸序列同一性百分比的比对可以以本领域技术范围内的各种方式实现,例如,使用公众可获得的计算机软件,例如blast、blast-2、align或megalign(dnastar)软件。本领域技术人员可以确定用于比对序列的适当参数,包括在所比较的序列的全长上实现最大比对所需的任何算法。然而,为了本文的目的,使用序列比较计算机程序align-2产生氨基酸序列同一性百分比的值。align-2序列比较计算机程序由genentech,inc授权,并且源代码已经随用户文档提交至华盛顿特区20559的美国版权局,在那里它以美国版权登记号txu510087登记。align-2程序可从加利福尼亚州南旧金山的genentech公司公开获得,或者可以从源代码编译。应编译align-2程序以在包括数字unixv4.0d的unix操作系统上使用。所有序列比较参数均由align-2程序设置,不会变化。
在使用align-2进行氨基酸序列比较的情况下,给定氨基酸序列a与、和或者相对于给定氨基酸序列b的氨基酸序列同一性百分比(或者可以表述为给定的氨基酸序列a具有或者包含与、和或者相对于给定氨基酸序列b的一定值的氨基酸序列同一性百分比),如下计算:
100*(x/y)
其中x是序列比对程序align-2在该程序的a和b的比对中得到的相同匹配的氨基酸残基数,其中y是b中氨基酸残基的总数。可以理解,氨基酸序列a的长度不等于氨基酸序列b的长度,a与b的氨基酸序列同一性百分比不等于b与a的氨基酸序列同一性百分比。除非另有说明,否则本文中使用的全部氨基酸序列同一性百分比值通过使用如前一段中所述的align-2计算机程序获得。
本文中使用的术语“药物制剂”是指这样的形式的制剂,其允许其中包含的活性成分的生物活性有效,并且不含有对施用所述制剂的个体具有无法接受的毒性的其他成分。
本文中使用的术语“药学上可接受的载体”是指药物制剂中除活性成分外的对个体无毒的成分。药学上可接受的载体包括但不限于缓冲剂、赋形剂、稳定剂或防腐剂。
本文中使用的术语“纯化的”和“分离的”是指根据本发明的抗体或核苷酸序列,所述分子在基本上不存在相同类型的其他生物大分子的情况下存在。本文中所用的术语“纯化的”优选是指存在至少75重量%,更优选至少85重量%,还更优选至少95重量%,最优选至少98重量%的相同类型的生物大分子。编码特定多肽的“分离的”核酸分子是指基本上不含不编码多肽的其他核酸分子的核酸分子;然而,该分子可包括一些不会有害地影响组合物基本特征的某些其它碱基或部分。
本文中使用的术语“重组抗体”是指由重组宿主细胞表达的抗体(例如嵌合、人源化或人抗体或其抗原结合片段),所述重组宿主细胞包含编码该抗体的核酸。用于制备重组抗体的“宿主细胞”的实例包括:(1)哺乳动物细胞,例如中国仓鼠卵巢(cho)、cos、骨髓瘤细胞(包括y0和ns0细胞)、幼仓鼠肾(bhk)、hela和vero细胞;(2)昆虫细胞,例如sf9,sf21和tn5;(3)植物细胞,例如属于烟草属的植物(烟草(nicotianatabacum));(4)酵母细胞,例如属于酵母属(saccharomyces)的酵母细胞(例如酿酒酵母(saccharomycescerevisiae))或属于曲霉属的酵母细胞(例如黑曲霉(aspergillusniger));(5)细菌细胞,例如大肠杆菌(escherichia.coli)细胞或枯草芽孢杆菌(bacillussubtilis)细胞等。
术语本发明的抗体或抗体片段的“治疗有效量”是指以适用于任何医学治疗的合理益处/风险比下用以治疗疾病或疾患的足够量的抗体或抗体片段。然而,应该理解,本发明的抗体或抗体片段和组合物的每日总用量将由主治医师在合理的医学判断范围内决定。任何特定患者的特定治疗有效剂量水平将取决于多种因素,包括所治疗的疾病和疾病的严重程度;所用特异性抗体或抗体片段的活性;使用的具体组合物,患者的年龄、体重、一般健康状况、性别和饮食;施用时间、施用途径和所用特异性抗体或抗体片段的排泄率;治疗的持续时间;与所用特定抗体组合使用或同时使用的药物;以及医学领域众所周知的因素。例如,本领域技术人员众所周知的是,以低于实现所需治疗效果所需的剂量水平开始用药,并逐渐增加剂量直至达到所需效果。
本文中使用的术语“单链fv”(“scfv”)是共价连接的vh::vl异二聚体,其通常由基因融合体表达,所述基因融合体包括由编码肽的接头连接的vh和vl编码基因。“dsfv”是通过二硫键稳定的vh::vl异二聚体。二价和多价抗体片段可以通过单价scfv的结合自发形成,或者可以通过肽接头偶联单价scfv产生,例如二价sc(fv)2。
本文中使用的术语“疗法”,“治疗”或“处理”是指试图改变所治疗的个体的自然病程的临床干预,并且可以用于预防或在临床病理过程中进行。理想的治疗效果包括但不限于预防疾病的发生或复发,症状的缓解,疾病的任何直接或间接病理后果的减少,预防转移,降低疾病进展的速度,改善或缓解疾病状态,缓解或改善预后。在一些实施方案中,本发明的抗体或抗体片段用于延迟疾病的发展或减缓疾病的进展。
本文中使用的术语“肿瘤”是指所有肿瘤细胞的生长和增殖,无论是恶性的还是良性的,以及所有癌前的及癌性的细胞和组织的生长和增生。本文中提及的术语“癌症”,“癌性”,“细胞增生性疾病”,“增生性疾病”和“肿瘤”不是相互排斥的。
本文中使用的术语“可变区”或“可变结构域”是指抗体重链或轻链的参与抗体与抗原的结合的结构域。天然抗体的重链和轻链(分别为vh和vl)的可变结构域通常具有相似的结构,每个结构域包含四个保守框架区(fr)和三个高变区(hvr)(参见,例如,kindt等,kubyimmunology,第6版,w.h.freemanandco.,第91页(2007))。单个vh或vl结构域可足以赋予抗原结合特异性。此外,可以使用来自结合特定抗原的抗体的vh或vl结构域以分别筛选互补vl或vh结构域的文库,从而分离结合该抗原的抗体或抗体片段。参见,例如,portolano等,j.immunol.,第150卷,第880-887页,1993;clarkson等,nature,第352卷,第624-628页,1991。
本文中使用的术语“载体”是指能够扩增与其连接的另一核酸的核酸分子。该术语包括作为自我复制核酸结构的载体以及掺入其已被引入的宿主细胞的基因组中的载体。某些载体能够指导它们可操作地连接的核酸的表达。此类载体在本文中称为“表达载体”。
具体实施方式
为了说明的目的,通过参考各种示例性实施方案来描述本发明的原理。虽然本文具体描述了本发明的某些实施方案,但是本领域普通技术人员将容易理解,相同的原理同样适用于其他系统和方法,并且可以用于其他系统和方法中。在详细解释本发明的公开实施方案之前,应该理解,本发明不限于其应用于任何特定实施方案所示的细节。另外,本文使用的术语是出于描述的目的而不是限制性的。此外,尽管参考本文以特定顺序呈现的步骤描述了某些方法,但在许多情况下,这些步骤可以以本领域技术人员可以理解的任何顺序执行;因此,新的方法不限于本文公开的步骤的特定排列。
必须注意,如本文和所附权利要求中所使用的,除非上下文另有明确说明,单数形式“一”、“一个”和“该”包括复数指代。此外,术语“一”(或“一个”)、“一个或多个”和“至少一个”在本文中可互换使用。术语“包含”,“包括”,“具有”和“由......构成”也可以互换使用。
除非另有说明,否则在说明书和权利要求中使用的表示成分的量,表示性质例如分子量、百分比、比值、反应条件等的所有数字应理解为在所有情况下都被术语“约”修改,而无论是否存在术语“约”。因此,除非有相反的指示,否则说明书和权利要求书中列出的数值参数是近似值,其可以根据本发明的所寻求获得的所需性质而变化。至少,并不是试图将等同原则的应用限制在权利要求的范围内,每个数值参数至少应该根据报告的有效数字的数量并通过应用普通的舍入技术来解释。尽管说明本发明广泛范围的数值范围和参数是近似值,但是在具体实施例中尽可能精确地报告列出的数值。然而,任何数值固有地包含必然由其各自的测试测量中存在的标准偏差引起的某些误差。
应理解,本文公开的每种组分、化合物、取代基或参数应被解释为单独使用或与本文公开的每种/每一其他组分、化合物、取代基或参数中的一种或多种组合使用。
还应理解,本文公开的每种组分、化合物、取代基或参数的每个量/值或者量/值的范围应解释为还与本文公开的任何其他组分、化合物、取代基或参数的每个量/值或者量/值的范围组合公开,因此,为了本发明的目的,本文公开的两种或更多种组分、化合物、取代基或参数的量/值或量/值范围的任何组合也因此彼此组合地公开。
还应理解,本文公开的每个范围的每个下限应解释为与本文公开的相同组分、化合物、取代基或参数的每个范围的每个上限组合公开。因此,两个范围的公开将被解释为通过将每个范围的每个下限与每个范围的每个上限组合而得到的四个范围的公开。三个范围的公开被解释为通过将每个范围的每个下限与每个范围的每个上限组合而得到的九个范围的公开,等等。此外,在说明书或实施例中公开的组分、化合物、取代基或参数的具体量/值被解释为公开范围的下限或上限,因此,可以与本申请中其他处公开的相同组分、化合物、取代基或参数的范围或特定量/值的任何其他下限或上限组合,以形成该组分、化合物、取代基或参数的范围。
a.抗axl抗体或抗体片段的区
一方面,本发明提供了分离的重链可变区多肽,其特异性结合人axl蛋白。重链可变区多肽包含三个互补决定区h1、h2和h3序列,其中:
h1序列为x1gx2x3mx4(seqidno:1);
h2序列为likx5snggtx6ynqkfkg(seqidno:2);
h3序列为gx7x8x9x10x11x12x13x14dyx15x16(seqidno:3),
其中,
x1为t或a或w,
x2为h或a,
x3为t或i,
x4为n或i,
x5为p或n,
x6为s或i或t,
x7为h或d或e或p或r或w,
x8为y或n,
x9为e或a或d或f或g或h或i或l或m或n或r或v或y,
x10为s或d或m或n或q,
x11为y或c或e或p,
x12为f或e或n或s或t或v,
x13为a或d或g或l或y,
x14为m或e或f,
x15为w或a或d或h或l或n或p或r或t,
x16为g或h。
重链可变区的比对显示于图1a中,其中互补决定区h1、h2和h3被框出。
另一方面,本发明提供了一种分离的轻链可变区多肽,其特异性结合人axl蛋白。轻链可变区多肽包含三个互补决定区l1、l2和l3序列,其中:
l1序列为kasqdx17x18sx19vx20(seqidno:4);
l2序列为x21x22x23trx24t(seqidno:5);
l3序列为qex25x26sx27x28x29x30(seqidno:6),
其中,
x17为v或d或g或n或w,
x18为s或v,
x19为a或l或m,
x20为a或d或n或q,
x21为w或f,
x22为a或i或n或p或q,
x23为s或d,
x24为h或d,
x25为h或c或f或i或l或q或s或t或v或y,
x26为f或c或d或e或g或n或s,
x27为t或c或p,
x28为p或a或c或d或e或h或k或s或t或v或w,
x29为l或g或r,
x30为t或i或r。
轻链可变区的比对显示在图1b中,其中互补决定区l1、l2和l3被框出。
本发明使用美国第8,709,755号专利中公开的方法从亲本抗体中鉴定了这些分离的重链可变区多肽和分离的轻链可变区多肽。亲本抗体(063-hum10f10)的重链可变区和轻链可变区也在图1a-1b中进行了比对,以示出分离的重链可变区多肽和分离的轻链可变区多肽中的突变。
使用综合位置演化(cpe)使编码野生型抗体的dna演化以产生突变抗体文库,其中模板抗体中的每个位置一次一个地随机化突变。文库中的每个突变抗体仅具有一个单点突变。通过elisa,在ph6.0和ph7.4下同时筛选对axl的选择性结合亲和力,产生文库中的突变抗体。使用两种突变抗体稀释液:1:3稀释液和1:9稀释液。选择在1:3稀释或1:9稀释的条件下在ph6.0下的结合亲和力与ph7.4下的结合亲和力的比值至少为1.5的突变抗体作为条件活性抗体,单点突变在每个重链可变区和轻链可变区中示出(表1和2)。
表1:条件活性抗axl抗体重链可变区
表2:条件活性抗axl抗体轻链可变区
在另一方面,本发明鉴定了如图1a中所示的重链可变区和如图1b中所示的轻链可变区。一些重链可变区由具有seqidno:11-13的dna序列编码。一些轻链可变区由具有seqidno:7-10的dna序列编码。这些重链可变区和轻链可变区可以特异性结合axl。已发现包含这些重链可变区和轻链可变区之一的抗体在肿瘤微环境中的ph下比在非肿瘤微环境中的ph下对ax1具有更高的结合亲和力。
本发明还包括图1a-1b中所示的重链可变区和轻链可变区的变体,所述变体由具有seqidno:9-13的dna序列编码,可以特异性结合axl。为了获得这些变体,决定了重链可变区(h1-h3)的互补决定区(cdr)和轻链可变区(l1-l3)的cdr应保持完整。
在获取这些变体时,可以依据本文所述的方法。这些重链可变区和轻链可变区的变体可以通过在编码重链可变区和轻链可变区的核苷酸序列中引入适当的修饰来制备,或通过肽合成来制备。此类修饰包括例如抗体或抗体片段的氨基酸序列内的残基的缺失、和/或插入和/或取代。可以采用缺失、插入和取代的任何组合来获得最终构建体,只要最终构建体具有至少一种所需特征,例如抗原结合。
取代、插入和缺失变体
在某些实施方案中,提供了具有一个或多个氨基酸取代的抗体或抗体片段变体。用于取代性诱变的目的位点包括cdr和框架区(fr)。在“保守性取代”的标题下表3中示出了保守性取代。并且如下文参考氨基酸侧链类别所进一步描述的,表3中在“示例性取代”的标题下提供了更多的取代性变化。可以将氨基酸取代引入目的抗体或抗体片段中,并筛选产物以获得所需的活性,例如保留/改善的抗原结合,或降低的免疫原性。
表3:氨基酸取代
氨基酸可根据常见的侧链性质分组:
(1)疏水性:正亮氨酸、met、ala、val、leu、ile;
(2)中性亲水:cys、ser、thr、asn、gln;
(3)酸性:asp、glu;
(4)碱性:his、lys、arg;
(5)影响链取向的残基:gly、pro;
(6)芳香族:trp、tyr、phe。
非保守性取代将需要将这些类别之一的成员替换为另一类别。
一种类型的取代变体涉及取代亲本抗体(例如人源化或人抗体)的一个或多个高变区残基。通常,为进一步研究而选择的所得变体,相对于亲本抗体具有某些生物学特性(例如,增加的亲和力、降低的免疫原性)方面有变化(例如,改进)和/或基本上保留了亲本抗体的某些生物学特性。示例性的取代变体是亲和力成熟的抗体,其可以例如使用如本文所述的基于噬菌体展示的亲和力成熟技术方便地产生。简而言之,使一个或多个cdr残基突变,在噬菌体上展示抗体变体并筛选具有特定的生物活性(例如结合亲和力)的抗体变体。
可以在cdr中进行改变(例如,取代),以改善例如抗体亲和力。这种改变可以发生在cdr“热点”,即由在体细胞成熟过程中以高频率发生突变的密码子编码的残基(参见,例如,chowdhury,methodsmol.biol.,第207卷,第179-196页,2008),和/或sdr(a-cdr),测试所得变体vh或vl的结合亲和力。通过构建二级文库和从二级文库中再筛选进行的亲和力成熟已经描述于例如hoogenboom等,methodsinmolecularbiology,第178卷,第1-37页,2001。在亲和力成熟的一些实施方案中,通过多种方法中的任何一种(例如,易错pcr,链改组或寡核苷酸定向诱变)将多样性引入所选择的用于进行成熟的可变基因中。然后创建二级文库。然后筛选该文库以鉴定具有所需亲和力的任何抗体变体。引入多样性的另一种方法涉及cdr定向方法,其中随机化突变几个cdr残基(例如,每次4-6个残基)。参与抗原结合的cdr残基可以例如使用丙氨酸扫描诱变或建模来明确地鉴定。特别是通常靶向cdr-h3和cdr-l3。
在某些实施方案中,取代、插入或缺失可以在一个或多个hvr内发生,只要这种改变不会实质上降低抗体或抗体片段结合抗原的能力。例如,可以在cdr中进行不会实质上降低结合亲和力的保守性改变(例如,如本文提供的保守性取代)。这种改变可在cdr“热点”或sdr之外。在上文提供的vh和vl序列变体的某些实施方案中,每个cdr均未被改变,或含有不超过一个、两个或三个氨基酸取代。
用于鉴定可以用作诱变的靶标的的抗体残基或区域的方法称为“丙氨酸扫描诱变”,描述于cunningham和wells,science,第244卷,第1081-1085页,1989。在该方法中,鉴定残基或一组靶残基(例如带电残基,例如arg、asp、his、lys和glu),并用中性或带负电荷的氨基酸(例如,丙氨酸或聚丙氨酸)取代,以确定抗体或抗体片段与抗原的相互作用是否受到影响。可以在对初始取代显示出功能敏感性的氨基酸位置引入进一步的取代。或者或并且,抗原-抗体复合物的晶体结构用于鉴定抗体或抗体片段与抗原之间的接触点。可以靶向或消除这些接触残基和邻近的残基作为取代的候选项。可以筛选变体以确定它们是否具有所需的性质。
氨基酸序列插入包括氨基-和/或羧基-末端融合,长度范围从一个残基到含有一百个或更多个残基的多肽,以及单个或多个氨基酸残基的序列内插入。末端插入的实例包括具有n-末端甲硫氨酰残基的抗体。抗体的其他插入变体包括抗体的n-或c-末端与酶(例如用于adept)或增加抗体血清半衰期的多肽的融合。
考虑了本文描述的抗体的氨基酸序列修饰。例如,可能需要改善抗体的结合亲和力和/或其他生物学特性。已知当通过将来源于非人动物的抗体的vh和vl中的cdr简单地移植到人抗体的vh和vl的fr中来制备人源化抗体时,与源自非人动物的原始抗体的抗原结合活性相比,其抗原结合活性降低。考虑到非人抗体的vh和vl不仅在cdr中而且在fr中均有几个氨基酸残基与抗原结合活性直接或间接相关。因此,用源自人抗体的vh和vl的fr的不同氨基酸残基取代这些氨基酸残基会降低结合活性。为了解决该问题,在移植有人cdr的抗体中,必须尝试在人抗体的vh和vl的fr的氨基酸序列中鉴定与抗体结合直接相关的氨基酸残基,或者与cdr的氨基酸残基相互作用的氨基酸残基,或者维持抗体的三维结构并且与抗原结合直接相关的氨基酸残基。通过用来源于非人动物的原始抗体的氨基酸残基替换所鉴定的氨基酸,可以提高所述被降低的抗原结合活性。
可以对本发明的抗体的结构和编码它们的dna序列进行修饰和改变,而仍然获得编码具有所需特征的抗体的功能分子。
在对进行氨基酸序列的改变时,可以考虑氨基酸的亲水指数。本领域通常理解亲水氨基酸指数在赋予蛋白交互生物学功能方面的重要性。人们认为氨基酸的相对亲水特性有助于所得蛋白的二级结构,该二级结构又决定了蛋白与其他分子例如酶、底物、受体、dna、抗体、抗原等的相互作用。以氨基酸的疏水性和电荷特征为基础,为各种氨基酸指定亲水指数,它们是:异亮氨酸(+4.5)、缬氨酸(+4.2)、亮氨酸(+3.8)、苯丙氨酸(+2.8)、半胱氨酸/胱氨酸(+2.5)、蛋氨酸(+1.9)、丙氨酸(+1.8)、甘氨酸(-0.4)、苏氨酸(-0.7)、丝氨酸(-0.8)、色氨酸(-0.9)、酪氨酸(-1.3)、脯氨酸(-1.6)、组氨酸(-3.2)、谷氨酸(-3.5)、谷氨酰胺(-3.5)、天冬氨酸(-3.5)、天冬酰胺(-3.5)、赖氨酸(-3.9)和精氨酸(-4.5)。
本发明的另一个目的还包括本发明抗体的功能保守性变体。
“功能保守性变体”“是其中蛋白或酶中的给定氨基酸残基已被改变而不改变多肽的整体构象和功能的变体,包括但不限于用具有相似性质(例如,极性,氢键电位,酸性,碱性,疏水性,芳香性等)的氨基酸替换氨基酸。除了被标示为是保守性的那些氨基酸之外,氨基酸可以在蛋白上不同,使得具有相似功能的任何两种蛋白之间的蛋白或氨基酸序列相似性百分比可以变化,例如根据诸如通过聚类方法(clustermethod)的比对方案测定的序列相似性百分比可以是70%至99%,其中相似性基于megalign算法。“功能保守性变体”还包括通过blast或fasta算法测定为具有至少60%氨基酸同一性的多肽,优选具有至少75%,更优选具有至少85%,又更优选具有至少90%,还更优选具有至少95%氨基酸同一性的多肽,并且所述多肽具有与其所比较的天然蛋白或亲本蛋白相同或基本相似的性质或功能。
当超过80%,优选超过85%,优选超过90%的氨基酸相同时,或者在较短序列的整个长度上超过约90%,优选超过95%是相似的(功能相同)时,两个氨基酸序列是“基本上同源的”或“基本上相似的”。优选使用例如gcg(geneticscomputergroup,programmanualforthegcgpackage,第7版,madison,wis.)pileup程序或任何序列比较算法(例如blast、fasta等)通过比对鉴定相似序列或同源序列。
例如,在蛋白结构中,某些氨基酸可以被其他氨基酸取代而没有明显的活性丧失。由于蛋白的相互作用能力和蛋白的性质决定了蛋白的生物功能活性,因此可以在蛋白序列中进行某些氨基酸取代,当然,也可以在其dna编码序列中进行,同时获得具有相似性质的蛋白。因此预期可以对本发明的抗体或抗体片段的序列或编码所述抗体或抗体片段的相应dna序列进行各种改变,而不会明显丧失其生物活性。
本领域已知某些氨基酸可以被具有相似的亲水指数或分值的其他氨基酸取代,并且仍然产生具有相似生物活性的蛋白,即仍然获得生物学功能等同的蛋白。
如上所述,氨基酸取代因此通常基于氨基酸侧链取代基的相对相似性,例如,它们的疏水性、亲水性、电荷、大小等。考虑到各种前述特征的示例性取代是本领域技术人员公知的,包括:精氨酸和赖氨酸;谷氨酸和天冬氨酸;丝氨酸和苏氨酸;谷氨酰胺和天冬酰胺;以及缬氨酸、亮氨酸和异亮氨酸。
糖基化变体
在某些实施方案中,改变本文提供的抗体以增加或降低抗体糖基化的程度。向抗体添加糖基化位点或缺失糖基化位点可以通过改变氨基酸序列使得产生一个或多个糖基化位点或者除去一个或多个糖基化位点而方便地完成。
在抗体包含fc区的情况下,可以改变与其连接的碳水化合物。由哺乳动物细胞产生的天然抗体通常包含支链的双触角寡糖,其通常通过n-键连接至fc区的ch2结构域的asn297。参见,例如,wright等,tibtech,第15卷,第26-32页,1997。寡糖可包括各种碳水化合物,例如甘露糖、n-乙酰葡糖胺(glcnac)、半乳糖、唾液酸、以及连接至双触角寡糖结构“主干”中的glcnac的海藻糖。在一些实施方案中,可以对本发明的抗体中的寡糖进行修饰以产生具有某些改善的性质的抗体变体。
在一个实施方案中,提供了具有碳水化合物结构的抗体变体,所述碳水化合物结构缺乏(直接或间接)连接至fc区的海藻糖。例如,这种抗体中海藻糖的量可以是1%至80%,1%至65%,5%至65%,或20%至40%。海藻糖的量通过计算asn297处糖链内海藻糖相对于与asn297连接的所有糖结构的总和(例如复合物,杂交和高甘露糖结构)的平均量来确定,如通过诸如wo2008/077546中所述的maldi-tof质谱法所测量的,例如,asn297是指位于fc区的约297位(fc区残基的eu编号)的天冬酰胺残基;然而,由于抗体中的微小序列变异,asn297也可位于297位上游或下游的约±3个氨基酸处,即294位和300位之间。此类海藻糖基化变体可具有改善的adcc功能。参见,例如,美国第us2003/0157108号(presta,l.)专利公开,和us2004/0093621(kyowahakkokogyoco.,ltd)。与“去海藻糖基化”或“海藻糖不足”的抗体变体相关的出版物的实例包括:us2003/0157108;wo2000/61739;wo2001/29246;us2003/0115614;us2002/0164328;us2004/0093621;us2004/0132140;us2004/0110704;us2004/0110282;us2004/0109865;wo2003/085119;wo2003/084570;wo2005/035586;wo2005/035778;wo2005/053742;wo2002/031140;okazaki等,j.mol.biol.,第336卷,第1239-1249页,2004;yamane-ohnuki等,biotech.bioeng.,第87卷,第614-622页,2004。能够产生去海藻糖基化抗体的细胞系的实例包括在蛋白海藻糖基化方面有缺陷的lec13cho细胞(ripka等,arch.biochem.biophys.,第249卷,第533-545页,1986;美国第us2003/0157108a号专利申请;和wo2004/056312a1,特别是实施例11),和敲除细胞系,例如α-1,6-海藻糖基转移酶基因、fut8、敲除的cho细胞(参见,例如,yamane-ohnuki等,biotech.bioeng.,第87卷,第614-622页,2004;kanda,y.等,biotechnol.bioeng.,第94卷,第680-688页,2006;和wo2003/085107)。
进一步提供具有二等分型寡糖的抗体变体,例如,其中连接于抗体fc区的双触角寡糖被glcnac一分为二。此类抗体变体可具有减少的海藻糖基化和/或改善的adcc功能。此类抗体变体的实例描述于例如wo2003/011878;美国专利第6,602,684号;和us2005/0123546。还提供了与fc区连接的寡糖中具有至少一个半乳糖残基的抗体变体。此类抗体变体可具有改善的cdc功能。此类抗体变体描述于例如wo1997/30087;wo1998/58964;和wo1999/22764。
fc区变体
在某些实施方案中,可以将一个或多个氨基酸修饰引入本文提供的抗体的fc区,从而产生fc区变体。fc区变体可包含在一个或多个氨基酸位置包含氨基酸修饰(例如取代)的人fc区序列(例如,人igg1、igg2、igg3或igg4fc区)。
在某些实施方案中,本发明考虑了具有部分但不是所有效应子功能的抗体变体,这使得所述抗体变体成为用于如下应用的理想候选物:在所述应用中,抗体体内半衰期很重要,但某些效应子功能(例如adcc)是不必要的或者是有害的。可以进行体外和/或体内细胞毒性测定以确认cdc和/或adcc活性的降低/消除。例如,可以进行fc受体(fcr)结合测定以确保抗体缺乏fcγr结合(因此可能缺乏adcc活性),但保留fcrn结合能力。介导adcc的原代细胞(nk细胞)仅表达fcγriii,而单核细胞表达fcγri、fcγrii和fcγriii。造血细胞上的fcr表达总结于ravetch和kinet,annu.rev.immunol.,第9卷,第457-492页,1991中的第464页表5。用于评估目标分子的adcc活性的体外试验的非限制性实例描述于美国第5,500,362号专利(也参见例如hellstrom等,proc.nat’lacad.sci.usa,第83卷,第7059-7063页,1986)和hellstrom,i等,proc.nat’lacad.sci.usa,第82卷,第1499-1502页,1985;美国第5,821,337号专利(也参见bruggemann等,j.exp.med.,第166卷,第1351-1361页,1987)。或者,可以使用非放射性测定方法,参见,例如,用于流式细胞术的actitm非放射性细胞毒性测定法(celltechnology,inc.mountainview,calif.);和
具有降低的效应子功能的抗体包括具有fc区残基238、265、269、270、297、327和329中的一个或多个残基的取代的抗体(美国第6,737,056号专利)。此类fc突变体包括在氨基酸位置265、269、270、297和327中的两个或更多个位置处具有取代的fc突变体,包括残基265和297取代为丙氨酸的所谓“dana”fc突变体(美国第7,332,581号专利)。
描述了与fcr的结合改善或减弱的某些抗体变体(参见,例如,美国第6,737,056号专利;wo2004/056312,和shields等,j.biol.chem.,第9卷,第6591-6604页,2001)。
在某些实施方案中,抗体变体包含具有改善adcc的一个或多个氨基酸取代的fc区,例如fc区的第298、333和/或334位处的取代(eu残基编号)。
在一些实施方案中,在fc区中进行改变,所述改变导致c1q结合和/或补体依赖性细胞毒性(cdc)的变化(即,提高或降低),例如,描述于美国第6,194,551号专利,wo99/51642,和idusogie等,j.immunol.,第164卷,第4178-4184页,2000。
在us2005/0014934中描述了具有增加的半衰期和改善的与新生儿fc受体(fcrn)结合的抗体,新生儿fc受体负责母体igg向胎儿的转移(guyer等,j.immunol.,第117卷,第587-593页,1976和kim等,j.immunol.,第24卷,第249页,1994)。那些抗体包含其中具有改善fc区与fcrn的结合的一个或多个取代的fc区。此类fc变体包括在fc区残基中的一个或多个残基处(238、256、265、272、286、303、305、307、311、312、317、340、356、360、362、376,378、380、382、413、424或434)具有取代的那些,例如,fc区残基434的取代(美国第7,371,826号专利)。涉及fc区变体的其他实例也可参见duncan&winter,nature,第322卷,第738-740页,1988;美国第5,648,260号专利;美国第5,624,821号专利;和wo94/29351。
半胱氨酸工程改造的抗体变体
在某些实施方案中,可能需要产生半胱氨酸工程改造的抗体,例如“thiomab”,其中抗体的一个或多个残基被半胱氨酸残基取代。在特定的实施方案中,取代的残基出现在抗体的可及位点。通过用半胱氨酸取代那些残基,反应性硫醇基团由此定位在抗体的可及位点,并且其可以用于将抗体与其他部分(例如药物部分或接头-药物部分)缀合,以产生如本文进一步描述的免疫缀合物。在某些实施方案中,以下残基中的任何一个或多个可以被半胱氨酸取代:轻链的v205(kabat编号);重链的a118(eu编号)和重链fc区的5400(eu编号)。可以按照例如美国第7,521,541号专利中所描述的来产生半胱氨酸工程改造的抗体。
抗体衍生物
在某些实施方案中,可进一步修饰本文提供的抗体或抗体片段以包含本领域已知且易于获得的其他非蛋白的部分。适于衍生化抗体或抗体片段的部分包括但不限于水溶性聚合物。水溶性聚合物的非限制性实例包括但不限于聚乙二醇(peg)、乙二醇/丙二醇的共聚物、羧甲基纤维素、葡聚糖、聚乙烯醇、聚乙烯吡咯烷酮、聚-1,3-二氧戊环、聚-1,3,6-三噁烷、乙烯/马来酸酐共聚物、聚氨基酸(均聚物或无规共聚物)、和葡聚糖或聚(n-乙烯基吡咯烷酮)聚乙二醇、丙二醇均聚物、环氧丙烷/环氧乙烷共聚物、聚氧乙烯化多元醇(例如甘油)、聚乙烯醇及其混合物。聚乙二醇丙醛由于其在水中的稳定性而在制备中具有优势。聚合物可以是任何分子量,并且可以是支链的或非支链的。与抗体或抗体片段连接的聚合物的数量可以变化,并且如果连接多于一种的聚合物,聚合物可以是相同的分子或不同的分子。通常,用于衍生化的聚合物的数量和/或类型可以基于考虑到的事项来确定,包括但不限于待改进的抗体或抗体片段的特定性质或功能,衍生物是否将用于在规定条件下的治疗等。
在另一个实施方案中,提供了抗体或抗体片段与可以通过暴露于辐射而被选择性加热的非蛋白部分的缀合物。在一个实施方案中,非蛋白部分是碳纳米管(kam等,proc.natl.acad.sci.usa,第102卷,第11600-11605页,2005)。辐射可以是任何波长的,包括但不限于不损害普通细胞但是将非蛋白部分加热到将抗体-非蛋白部分附近的细胞杀死的温度的波长。
另一方面,本发明提供了抗axl抗体或抗体片段,其包括分离的重链可变区多肽或分离的轻链可变区多肽。分离的重链可变区多肽包含分别具有seqidno:1-3的h1、h2和h3区。分离的轻链可变区多肽包含分别具有seqidno:4-6的l1、l2和l3区。
本发明的抗axl抗体或抗体片段在肿瘤微环境条件下比在非肿瘤微环境条件下对ax1具有更高的结合亲和力。在一个实施方案中,肿瘤微环境中的条件和非肿瘤微环境中的条件都是ph。因此,本发明的抗axl抗体或抗体片段可以在约5-6.8的ph下选择性地与ax1结合,但在正常生理环境中遇到的ph7.2-7.8下对ax1具有较低的结合亲和力。如实施例3-4所示,抗axl抗体或抗体片段在ph6.0下比在ph7.4下具有更高的结合亲和力。
在某些实施方案中,本发明的抗axl抗体或抗体片段在肿瘤微环境的条件下与axl的解离常数(kd)为约≤1μm,≤100nm,≤10nm,≤1nm,≤0.1nm,≤0.01nm,或≤0.001nm(例如10-8m或更低,或10-8m至10-13m,或10-9m至10-13m)。在一个实施方案中,抗体或抗体片段在肿瘤微环境中条件的一值下与axl的kd,与非肿瘤微环境中相同条件的不同值下的kd的比为至少约1.5:1,至少约2:1,至少约3:1,至少约4:1,至少约5:1,至少约6:1,至少约7:1,至少约8:1,至少约9:1,至少约10:1,至少约20:1,至少约30:1,至少约50:1,至少约70:1,或至少约100:1。
在一个实施方案中,使用如下所述的测定法通过用目的抗体的fab形式及其抗原进行的放射性标记的抗原结合测定(ria)测量kd。fab对抗原的溶液结合亲和力通过在存在未标记抗原的滴定系列的情况下,用最小浓度的(125i)-标记的抗原平衡fab,接着用抗-fab抗体-包被的板捕获结合的抗原来测量(参见,例如,chen等,j.mol.biol.293:865-881(1999))。为了确定测定的条件,将
根据另一个实施方案,kd是通过表面等离子共振测定法使用
本发明的抗axl抗体可以是嵌合抗体,人源化抗体或人抗体。在一个实施方案中,使用抗axl抗体片段,例如fv、fab、fab’、fab’-sh、scfv、双体抗体、三体抗体(triabody)、四体抗体(tetrabody)或f(ab’)2片段和由抗体片段形成的多特异性抗体。在另一个实施方案中,抗体是全长抗体,例如完整igg抗体或本文定义的其他抗体类别或同种型。对于特定抗体片段的综述,参见hudson等,nat.med.,第9卷,第129-134页,2003。对于scfv片段的综述,参见,例如,pluckthün,thepharmacologyofmonoclonalantibodies,第113卷,rosenburg和moore编辑,(springer-verlag,纽约),第269-315页(1994);还可参见wo93/16185;和美国第5,571,894号和第5,587,458号专利。对于包含拯救受体结合表位残基和具有提高的体内半衰期的fab和f(ab’)2片段的讨论,参见美国第5,869,046号专利。
本发明的双体抗体可以是二价的或双特异性的。关于双体抗体的实例参见,例如,ep404,097;wo1993/01161;hudson等,nat.med.9:129-134(2003);和hollinger等,proc.natl.acad.sci.usa,第90卷,第6444-6448页,1993。三体抗体和四体抗体的实例也描述于hudson等,nat.med.,第9卷,第129-134页,2003。
在一些实施方案中,本发明包括单结构域抗体片段,其包含抗体的全部或部分重链可变结构域或全部或部分轻链可变结构域。在某些实施方案中,单结构域抗体是人单结构域抗体(domantis,inc.,waltham,mass.;参见,例如,美国第6,248,516b1号专利)。
抗体片段可以通过不同技术制备,包括但不限于完整抗体的蛋白水解消化以及通过重组宿主细胞(例如大肠杆菌或噬菌体)产生,如本文所述。
在一些实施方案中,本发明的抗axl抗体可以是嵌合抗体。某些嵌合抗体描述于例如美国专利第4,816,567号;和morrison等,proc.natl.acad.sci.usa,第81卷,第6851-6855页,1984。在一个实例中,嵌合抗体包含非人可变区(例如,源自小鼠、大鼠、仓鼠、兔或例如猴的非人灵长类动物的可变区)和人类恒定区。在另一个实例中,嵌合抗体是抗体的类别或亚类已经相对于亲本抗体的种类或亚类发生变化的“类别转变”抗体。嵌合抗体包括其抗原结合片段。
在某些实施方案中,本发明的嵌合抗体是人源化抗体。通常,这种非人类抗体经过人源化以降低对人类的免疫原性,同时保持亲本非人类抗体的特异性和亲和力。通常,人源化抗体包含一个或多个可变结构域,其中cdr(或其部分)源自非人类抗体,fr(或其部分)源自人类抗体序列。人源化抗体还可任选地包含人类恒定区的至少一部分。在一些实施方案中,人源化抗体中的一些fr残基被来自非人类抗体(例如,cdr残基所源自的抗体)的相应残基取代,例如,以恢复或提高抗体特异性或亲和力。
人源化抗体及其制备方法于例如almagro和fransson,front.biosci.,第13卷,第1619-1633页,2008中有评述,且进一步描述于例如riechmann等,nature,第332卷,第323-329页,1988);queen等,proc.nat’lacad.sci.usa第86卷,第10029-10033页,1989;美国专利第5,821,337号、第7,527,791号、第6,982,321号和第7,087,409号;kashmiri等,methods,第36卷,第25-34页,2005(描述sdr(a-cdr)移植);padlan,mol.immunol.,第28卷,第489-498页,1991(描述“表面重整”);dall’acqua等,methods,第36卷,第43-60页,2005(描述“fr重组”);及osboum等,methods,第36卷,第61-68页,2005和klimka等,br.j.cancer,第83卷,第252-260页,2000(描述fr重组的“导向选择”方法)。
可用于人源化的人类框架区包括,但不限于,使用“最佳拟合”法选择的框架区(参见例如sims等,j.immunol.,第151卷,第2296页,1993);源自特定亚群的轻链或重链可变区的人类抗体的共有序列的框架区(参见例如carter等,proc.natl.acad.sci.usa,第89卷,第4285页,1992;及presta等,j.immunol.,第151卷,第2623页,1993);人类成熟(体细胞突变)框架区或人类生殖系框架区(参见例如almagro和fransson,front.biosci.,第13卷,第1619-1633页,2008);及源自筛选fr文库的框架区(参见例如baca等,j.biol.chem.,第272卷,第10678-10684页,1997及rosok等,j.biol.chem.,第271卷,第22611-22618页,1996)。
在一些实施方案中,本发明的抗axl抗体是多特异性的,例如,双特异性抗体。多特异性抗体是对至少两个不同位点具有结合特异性的单克隆抗体。在某些实施方案中,结合特异性之一针对ax1而另一种结合特异性针对另一种抗原。在某些实施方案中,双特异性抗体可以结合axl的两个不同表位。双特异性抗体也可用于将细胞毒性剂定位于表达axl的细胞。双特异性抗体可以制备成全长抗体或抗体片段。
制备多特异性抗体的技术包括但不限于具有不同特异性的两种免疫球蛋白重链-轻链对的重组共表达(参见milstein和cuello,nature,第305卷,第537-540页,1983),wo93/08829和traunecker等,emboj.,第10卷,第3655-3659页,1991)和“杵臼(knob-in-hole)”工程改造(参见例如美国专利第5,731,168号)。也可通过以下方法制得多特异性抗体:工程改造静电导引作用以制备抗体fc-异二聚体分子(wo2009/089004a1);将两种或两种以上抗体或片段交联(参见,例如,美国专利第4,676,980号,和brennan等,science,第229卷,第81-83页,1985);使用亮氨酸拉链来产生双特异性抗体(参见,例如,kostelny等,j.immunol.,第148卷,第1547-1553页,1992);使用“双体抗体”技术来制备双特异性抗体片段(参见,例如,hollinger等,proc.natl.acad.sci.usa,第90卷,第6444-6448页,1993);及使用单链fv(scfv)二聚体(参见例如gruber等,j.immunol.,第152卷,第5368-5374页,1994);及如tutt等,j.immunol.,第147卷,第60-69页,1991中所述制备三特异性抗体。
本文还包括具有三个或三个以上功能性抗原结合位点的经工程改造的抗体,包括“章鱼抗体”(参见例如us2006/0025576a1)。
抗体或抗体片段还可以包括包含结合至axl以及另一不同抗原的抗原结合位点的“双功能fab或“daf'”(例如参见us2008/0069820)。
可以使用重组方法和组合物产生本发明的抗axl抗体或抗体片段,其在us2016/0017040中有详细描述。
可以通过本领域已知的各种测定方法来测试和测量本发明的抗axl抗体或抗体片段的物理/化学性质和/或生物活性。这些测定方法中的一些方法描述于美国第8,853,369号专利中。
b.免疫缀合物
在另一个方面,本发明还提供免疫缀合物,其包含与一种或多种细胞毒性剂缀合的本文所述的抗axl抗体,所述细胞毒性剂例如化学治疗剂或药物、生长抑制剂、毒素(例如,蛋白毒素,细菌、真菌、植物或动物来源的酶活性毒素,或其片段)、或放射性同位素。
在一个实施方案中,免疫缀合物是抗体-药物缀合物(adc),其中抗体与一种或多种药物缀合,包括但不限于美登素类化合物(maytansinoid)(参见美国第5,208,020号和第5,416,064号专利和欧洲专利ep0425235b1);奥利斯他汀(auristatin)例如一甲基奥利斯他汀药物部分de和df(mmae和mmaf)(参见美国第5,635,483号、第5,780,588号和第7,498,298号专利);海兔毒素(dolastatin);卡奇霉素或其衍生物(参见美国第5,712,374号、第5,714,586号、第5,739,116号、第5,767,285号、第5,770,701号、第5,770,710号、第5,773,001号和第5,877,296号专利;hinman等,cancerres.,第53卷,第3336-3342页,1993;和lode等,cancerres.,第58卷,第2925-2928页,1998);蒽环类药物例如道诺霉素或多柔比星(参见kratz等,currentmed.chem.,第13卷,第477-523页,2006;jeffrey等,bioorganic&med.chem.letters,第16卷,第358-362页,2006;torgov等,bioconj.chem.,第16卷,第717-721页,2005;nagy等,proc.natl.acad.sci.usa,第97卷,第829-834页,2000;dubowchik等,bioorg.&med.chem.letters,第12卷,第1529-1532页,2002;king等,j.med.chem.,第45卷,第4336-4343页,2002;和美国第6,630,579号专利);甲氨蝶呤;长春地辛;紫杉烷例如多西紫杉醇、紫杉醇、拉洛他赛(larotaxel)、瞢司他赛(tesetaxel)和欧塔他赛(ortataxel);单端孢霉烯;和cc1065。
在另一实施方案中,免疫缀合物包含与酶活性毒素或其片段缀合的如本文所述的抗体,酶活性毒素或其片段包括,但不限于,白喉毒素a链、白喉毒素的非结合活性片段、外毒素a链(来自铜绿假单胞菌(pseudomonasaeruginosa))、蓖麻毒蛋白(ricin)a链、相思豆毒蛋白(abrin)a链、蒴莲根毒蛋白(modeccin)a链、α-帚曲霉素(sarcin)、油桐(aleutitesfordii)蛋白、香石竹毒蛋白(dianthinprotein)、美洲商陆(phytolacaamericana)蛋白(papi、papii和pap-s)、苦瓜(momordicacharantia)抑制剂、麻疯树毒蛋白(curcin)、巴豆毒蛋白(crotin)、肥皂草(sapaonariaofficinalis)抑制剂、白树毒蛋白(gelonin)、丝林霉素(mitogellin)、局限曲菌素(restrictocin)、酚霉素(phenomycin)、依诺霉素(enomycin)和单端孢霉烯族化合物(tricothecenes)。
在另一实施方案中,免疫缀合物包含如本文中所述的抗体与放射性原子缀合形成的放射性缀合物。多种放射性同位素可用于制备放射性缀合物。实例包括at211、i131、i125、y90、re186、re188、sm153、bi212、p32、pb212及lu的放射性同位素。当使用放射性缀合物进行检测时,其可包含用于闪烁摄影研究的放射性原子,例如tc99m或i123或用于核磁共振(nmr)成像(也称为磁共振成像,mri)的自旋标记物,例如碘-123及碘-131、铟-111、氟-19、碳-13、氮-15、氧-17、钆、锰。
抗体与细胞毒性剂的缀合物可使用多种双功能蛋白偶联剂制得,例如n-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(spdp)、琥珀酰亚胺基-4-(n-马来酰亚氨甲基)环己烷-1-羧酸酯(smcc)、亚氨基硫烷(it)、亚氨酸酯(例如盐酸二甲基己二亚酰胺化物)、活性酯类(例如辛二酸二琥珀酰亚胺基酯)、醛类(例如戊二醛)、双叠氮化合物(例如双(对-叠氮苯甲酰基)己二胺)、双重氮衍生物(例如双(对-重氮苯甲酰基)己二胺)、二异氰酸酯(例如甲苯2,6-二异氰酸酯)、和双活性氟化合物(例如1,5-二氟-2,4-二硝基苯)的双功能衍生物。举例来说,蓖麻毒蛋白免疫毒素可如vitetta等,science,第238卷,第1098页,1987中所述来制备。碳-14标记的1-异硫氰酸苯甲基-3-甲基二亚乙基三胺五乙酸(mx-dtpa)是用于使放射性核苷酸与抗体结合的示例性螯合剂。参见wo94/11026。接头可以是促进细胞毒性药物在细胞中释放的“可裂解接头”。举例来说,可使用酸不稳定接头、肽酶敏感性接头、光不稳定接头、二甲基接头或含二硫化物的接头(chari等,cancerres.,第52卷,第127-131页,1992;美国第5,208,020号专利)。
本文中的免疫缀合物明确涵盖但不限于用交联剂制备的缀合物,所述交联剂包括但不限于可商购(例如,来自piercebiotechnology,inc.,rockford,ill.,美国)的bmps、emcs、gmbs、hbvs、lc-smcc、mbs、mpbh、sbap、sia、slab、smcc、smpb、smph、硫代-emcs、硫代-gmbs、硫代-kmus、硫代-mbs、硫代-siab、硫代-smcc和硫代-smpb、以及svsb(琥珀酰亚胺基-(4-乙烯基砜)苯甲酸酯。
adc的示例性实施方案包括靶向肿瘤细胞的抗体(ab)、药物部分(d)和将ab连接至d的接头部分(l)。在一些实施方案中,抗体通过一个或多个氨基酸残基例如赖氨酸和/或半胱氨酸连接于接头部分(l)。
示例性adc具有为ab-(ld)p的式i,其中p为1至约20。在一些实施方案中,可以与抗体缀合的药物部分的数量受游离半胱氨酸残基数量的限制。在一些实施方案中,通过本文描述的方法将游离半胱氨酸残基引入抗体氨基酸序列中。式i的示例性adc包括但不限于具有1、2、3或4个工程改造的半胱氨酸氨基酸的抗体(lyon等,methodsinenzym.,第502卷,第123-138页,2012)。在一些实施方案中,一个或多个游离半胱氨酸残基已经存在于抗体中,而无需利用工程化改造,在这种情况下,现有的游离半胱氨酸残基可用于将抗体与药物缀合。在一些实施方案中,在缀合抗体之前将抗体暴露于还原条件,以产生一个或多个游离半胱氨酸残基。
a)示例性接头
“接头”(l)是双功能部分或多功能部分,其可用于将一个或多个部分(例如药物部分(d))与抗体(ab)连接以形成免疫缀合物,例如式i的adc。在一些实施方案中,可以使用具有用于共价连接至药物和抗体的反应性官能团的接头来制备adc。例如,在一些实施方案中,抗体(ab)的半胱氨酸硫醇基可以与接头的反应性官能团或药物-接头中间体形成键以制备adc。
在一个方面,接头具有能够与抗体上存在的游离半胱氨酸反应以形成共价键的官能团。非限制性的、示例性的这种反应性官能团包括马来酰亚胺、卤代乙酰胺、α-卤代乙酰基、活化酯类例如琥珀酰亚胺酯、4-硝基苯酯、五氟苯酯、四氟苯酯、酸酐、酰基氯、磺酰氯、异氰酸酯和异硫氰酸酯。参见,例如,klussman等的bioconjugatechemistry,第15卷,第765-773页,2004,第766页的缀合方法。
在一些实施方案中,接头具有能够与抗体上存在的亲电子基团反应的官能团。示例性的这种亲电子基团包括但不限于醛和酮羰基。在一些实施方案中,接头的反应性官能团的杂原子可以与抗体上的亲电子基团反应并与抗体单元形成共价键。非限制性的、示例性的这种反应性官能团包括但不限于酰肼、肟、氨基、肼、缩氨基硫脲、肼羧酸盐和芳基酰肼。
接头可包含一种或多种接头组分。示例性接头组分包括6-马来酰亚胺基己酰基(“mc”)、马来酰亚胺丙酰基(“mp”)、缬氨酸-瓜氨酸(“val-cit”或“vc”)、丙氨酸-苯丙氨酸(“ala-phe”)、对氨基苄氧基羰基(“pab”)、n-琥珀酰亚胺基-4-(2-吡啶硫基)戊酸酯(“spp”)和4-(n-马来酰亚胺甲基)环己烷-1-羧酸酯(“mcc”)。各种接头组分是本领域已知的,其中一些在下面描述。
接头可以是促进药物释放的“可切割的接头”。非限制性的示例性可切割的接头包括酸不稳定接头(例如,包含腙)、蛋白酶敏感的(例如,肽酶敏感的)接头、光不稳定接头或含二硫化物接头(chari等,cancerresearch,第52卷,第127-131页,1992;美国第5,208,020号专利)。
在某些实施方案中,接头具有式ii-aa-ww-yy-,其中a是“延伸单元”,a是0至1的整数;w是“氨基酸单元”,w是0至12的整数;y是“间隔物单元”,y是0、1或2。包含式ii的接头的adc具有式i(a):ab-(aa-ww-yy-d)p,其中ab、d和p如上式i所定义的那样。这种接头的示例性实施方案描述于美国第7,498,298号专利中。
在一些实施方案中,接头组分包含将抗体与另一个接头组分或药物部分连接的“延伸单元”(a)。非限制性的示例性延伸单元如下所示(其中波浪线表示与抗体、药物或其他接头组分共价连接的位点):
在一些实施方案中,接头组分包含“氨基酸单元”(w)。在一些这样的实施方案中,氨基酸单元允许由蛋白酶切割接头,从而在暴露于细胞内蛋白酶例如溶酶体酶时促进药物从免疫缀合物中释放(doronina等,nat.biotechnol.,第21卷,第778-784页,2003)。示例性的氨基酸单元包括但不限于二肽、三肽、四肽和五肽。示例性的二肽包括但不限于缬氨酸-瓜氨酸(vc或val-cit)、丙氨酸-苯丙氨酸(af或ala-phe);苯丙氨酸-赖氨酸(fk或phe-lys);苯丙氨酸-高赖氨酸(phe-homolys);和n-甲基-缬氨酸-瓜氨酸(me-val-cit)。示例性的三肽包括但不限于甘氨酸-缬氨酸-瓜氨酸(gly-val-cit)和甘氨酸-甘氨酸-甘氨酸(gly-gly-gly)。氨基酸单元可以包含天然存在的氨基酸残基和/或较小的氨基酸和/或非天然存在的氨基酸类似物,例如瓜氨酸。氨基酸单元可以被设计和优化用于由特定酶(例如,肿瘤相关蛋白酶,组织蛋白酶b、c和d,或纤溶酶蛋白酶)进行的酶促切割。
通常,肽型接头可以通过在两个或更多个氨基酸和/或肽片段之间形成肽键来制备。这种肽键可以例如根据液相合成方法制备(例如,e.schroder和k.lübke(1965)“thepeptides”,第1卷,第76-136页,academicpress)。
在一些实施方案中,接头组分包含“间隔物单元”(y),其直接或通过延伸单元和/或氨基酸单元将抗体与药物部分连接。间隔物单元可以是“自毁型的”或“非自毁型的”。“非自毁型的”间隔物单元是其中部分或全部间隔物单元在adc切割后保持与药物部分结合的单元。非自毁型的间隔物单元的实例包括但不限于甘氨酸间隔物单元和甘氨酸-甘氨酸间隔物单元。在一些实施方案中,通过肿瘤细胞相关蛋白酶酶促切割含有甘氨酸-甘氨酸间隔物单元的adc导致甘氨酸-甘氨酸-药物部分从adc的其余部分上释放出来。在一些这样的实施方案中,甘氨酸-甘氨酸-药物部分在肿瘤细胞中经历水解步骤,从而将药物部分和甘氨酸-甘氨酸间隔物单元切割开。
“自毁型的”间隔物单元允许释放药物部分。在某些实施方案中,接头的间隔物单元包含对-氨基苄基单元。在一些这样的实施方案中,对-氨基苄醇通过酰胺键与氨基酸单元连接,在苯甲醇和药物之间制备氨基甲酸酯、甲基氨基甲酸酯或碳酸酯(hamann等,expertopin.ther.patents,第15卷,第1087-1103页,2005)。在一些实施方案中,间隔物单元包含对-氨基苄氧基羰基(pab)。在一些实施方案中,包含自毁型的接头的adc具有以下结构:
其中q是-c1-c8烷基,-o-(c1-c8烷基),-卤素,-硝基或-氰基;m是0到4的整数;x可以是一个或多个其他的间隔物单元或可以不存在;p的范围为1至20。在一些实施方案中,p的范围为1至10、1至7、1至5或1至4。非限制性的示例性x间隔物单元包括:
其中r1和r2独立地选自h和c1-c6烷基。在一些实施方案中,r1和r2各自为-ch3。
自毁型的间隔物的其他实例包括但不限于在电子方面与pab基团相似的芳香族化合物,例如2-氨基咪唑-5-甲醇衍生物(美国第7,375,078号专利;hay等,bioorg.med.chem.lett.,第9卷,第2237页,1999)和邻-氨基苯基缩醛或对-氨基苯基缩醛。在一些实施方案中,可以使用在酰胺键水解时进行环化的间隔物,例如取代和未取代的4-氨基丁酸酰胺(rodrigues等,chemistrybiology,第2卷,第223页,1995),适当取代的双环[2.2.1]和双环[2.2.2]环系(storm等,j.amer.chem.soc.,第94卷,第5815页,1972)和2-氨基苯基丙酸酰胺(amsberry等,j.org.chem.,第55卷,第5867页,1990)。药物与甘氨酸残基的α-碳的连接是可用于adc的自毁型间隔物的另一个例子(kingsbury等,j.med.chem.,第27卷,第1447页,1984)。
在一些实施方案中,接头l可以是树枝状接头,其用于通过分支的多功能接头部分将一个以上的药物部分共价连接至抗体(sun等,bioorganic&medicinalchemistryletters,第12卷,第2213-2215页,2002;sun等,bioorganic&medicinalchemistry,第11卷,第1761-1768页,2003)。树枝状接头可以增加药物与抗体的摩尔比,即负载,其与adc的效能有关。因此,在抗体仅具有有一个反应性半胱氨酸硫醇基的情况下,可经由树枝状接头附接多个药物部分。
以下在式i的adc的情况下示出非限制性的示例性接头:
其中r1和r2独立地选自h和c1-c6烷基。在一些实施方案中,r1和r2各自为-ch3。
其中n为0至12。在一些实施方案中,n为2至10。在一些实施方案中,n为4至8。
其他非限制性的示例性adc包括以下结构:
y为:
每个r独立地为h或c1-c6烷基;n为1至12。
在一些实施方案中,接头被调节溶解度和/或反应性的基团取代。作为非限制性实例,带电荷的取代基例如磺酸根(-so3-)或铵基可以增加接头试剂的水溶性并促进接头试剂与抗体和/或药物部分的偶联反应,或者取决于用于制备adc的合成途径,促进ab-l(抗体-接头中间体)与d或者d-l(药物-接头中间体)与ab的偶联反应。在一些实施方案中,接头的一部分与抗体偶联,接头的一部分与药物偶联,然后ab-(接头部分)a与药物-(接头部分)b偶联以形成式i的adc。
本发明的化合物明确涵盖但不限于用以下接头试剂制备的adc:双马来酰亚胺基三氧乙二醇(bmpeo)、n-(β-马来酰亚胺基丙氧基)-n-羟基琥珀酰亚胺酯(bmps)、n-(ε-马来酰亚胺基己酰氧基)琥珀酰亚胺酯(emcs)、n-[γ-马来酰亚胺基丁酰氧基]琥珀酰亚胺酯(gmbs)、1,6-己烷-双-乙烯基砜(hbvs)、琥珀酰亚胺基-4-(n-马来酰亚胺甲基)环己烷-1-羧基-(6-氨基己酸酯)(lc-smcc)、间-马来酰亚胺基苯甲酰基-n-羟基琥珀酰亚胺酯(mbs)、4-(4-n-马来酰亚胺基苯基)丁酸酰肼(mpbh)、琥珀酰亚胺基3-(溴乙酰氨基)丙酸酯(sbap)、琥珀酰亚胺基碘乙酸酯(sia)、琥珀酰亚胺基(4-碘乙酰基)氨基苯甲酸酯(siab)、n-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(spdp)、n-琥珀酰亚胺基-4-(2-吡啶硫基)戊酸酯(spp)、琥珀酰亚胺基4-(n-马来酰亚胺甲基)环己烷-1-羧酸酯(smcc)、琥珀酰亚胺基4-(对-马来酰亚胺基苯基)丁酸酯(smpb)、琥珀酰亚胺基6-[(β-马来酰亚胺丙酰胺基)己酸酯](smph)、亚氨基硫烷(it)、硫代-emcs、硫代-gmbs、硫代-kmus、硫代mbs、硫代-siab、硫代-smcc和硫代-smpb、以及琥珀酰亚胺基-(4-乙烯基砜)苯甲酸酯(svsb),包括双-马来酰亚胺试剂:二硫代双马来酰亚胺乙烷(dtme)、1,4-双马来酰亚胺丁烷(bmb)、1,4-双马来酰亚胺基-2,3-二羟基丁烷(bmdb)、双马来酰亚胺基己烷(bmh)、双马来酰亚胺基乙烷(bmoe)、bm(peg)2(如下所示)和bm(peg)3(如下所示);亚氨酸酯的双功能衍生物(例如二亚胺代己二酸二甲酯盐酸盐)、活性酯(例如辛二酸二琥珀酰亚胺)、醛类(例如戊二醛)、双叠氮基化合物(例如双(对-叠氮基苯甲酰基)己二胺)、双重氮基衍生物(例如双-(对-重氮基苯甲酰基)-乙二胺)、二异氰酸酯(例如甲苯2,6-二异氰酸酯)和双活性氟化合物(例如1,5-二氟-2,4-二硝基苯)。在一些实施方案中,双马来酰亚胺允许抗体中半胱氨酸的硫醇基团连接至含硫醇基的药物部分、接头或接头-药物中间体。与硫醇基反应的其他官能团包括但不限于碘乙酰胺、溴乙酰胺、乙烯基吡啶、二硫化物、二硫化吡啶、异氰酸酯和异硫氰酸酯。
某些有用的接头试剂可以从各种商业来源获得,例如piercebiotechnology,inc.(rockford,ill.),molecularbiosciencesinc.(科罗拉多州,博尔德),或者根据本领域描述的方法合成;例如,toki等,j.org.chem.,第67卷,第1866-1872页,2002;dubowchik等,tetrahedronletters,第38卷,第5257-60页,1997;walker,j.org.chem.,第60卷,第5352-5355页,1995;frisch等,bioconjugatechem.,第7卷,第180-186页,1995;美国专利第6,214,345号;wo02/088172;us2003130189;us2003096743;wo03/026577;wo03/043583;和wo04/032828。
碳-14-标记的1-异硫氰酸基苄基-3-甲基二亚乙基三胺五乙酸(mx-dtpa)是用于将放射性核苷酸与抗体缀合的示例性螯合剂。参见,例如,wo94/11026。
b)示例性药物部分
1)美登素和美登素类化合物
在一些实施方案中,免疫缀合物包含与一种或多种美登素类化合物分子缀合的抗体。美登素类化合物是美登素的衍生物,是通过抑制微管蛋白聚合发挥作用的有丝分裂抑制剂。美登素最先从东非灌木齿叶美登木(maytenusserrata)中分离出来(美国第3,896,111号专利)。随后,发现某些微生物也产生美登素类化合物,例如美登醇和c-3美登醇酯(美国第4,151,042号专利)。合成的美登素类化合物公开于例如美国第4,137,230号专利;美国第4,248,870号专利;美国第4,256,746号专利;美国第4,260,608号专利;美国第4,265,814号专利;美国第4,294,757号专利;美国第4,307,016号专利;美国第4,308,268号专利;美国第4,308,269号专利;美国第4,309,428号专利;美国第4,313,946号专利;美国第4,315,929号专利;美国第4,317,821号专利;美国第4,322,348号专利;美国第4,331,598号专利;美国第4,361,650号专利;美国第4,364,866号专利;美国第4,424,219号专利;美国第4,450,254号专利;美国第4,362,663号专利;和美国第4,371,533号专利。
在抗体-药物缀合物中,美登素类化合物药物部分是有吸引力的药物部分,因为它们:(i)通过发酵或者化学修饰或衍生发酵产物而相对容易地制备,(ii)易于用适于通过非二硫化物接头与抗体缀合的官能团进行衍生化,(iii)在血浆中稳定,和(iv)有效对抗多种肿瘤细胞系。
适合用作美登素类化合物药物部分的某些美登素类化合物是本领域已知的,可以根据已知方法从天然来源分离或使用基因工程技术产生(参见,例如,yu等,pnas,第99卷,第7968-7973页,2002)。美登素类化合物也可以根据已知方法合成制备。
示例性美登素类化合物药物部分包括但不限于具有修饰的芳香环的那些,例如:c-19-脱氯(美国第4,256,746号专利)(例如通过氢化铝锂还原安丝菌素(ansamytocin)p2制备);c-20-羟基(或c-20-脱甲基)+/-c-19-脱氯(美国第4,361,650和第4,307,016号专利)(例如,通过使用链霉菌(streptomyces)或放线菌(actinomyces)进行脱甲基化或使用lah进行脱氯制备);和c-20-脱甲氧基,c-20-酰氧基(-ocor),+/-脱氯(美国第4,294,757号专利)(例如,通过使用酰氯进行酰化制备),以及在芳香环的其它位置处具有修饰的那些。
示例性美登素类化合物药物部分还包括具有如下修饰的那些,例如:c-9-sh(美国第4,424,219号专利)(例如,通过美登醇与h2s或p2s5的反应制备);c-14-烷氧基甲基(脱甲氧基/ch2or)(美国第4,331,598号专利);c-14-羟甲基或酰氧基甲基(ch2oh或ch2oac)(美国第4,450,254号专利)(例如,由诺卡氏菌(nocardia)制备);c-15-羟基/酰氧基(美国第4,364,866号专利)(例如,通过链霉菌转化美登醇制备);c-15-甲氧基(美国第4,313,946号和第4,315,929号专利)(例如,从滑桃树(trewianudlflora)分离);c-18-n-脱甲基(美国第4,362,663号和第4,322,348号专利)(例如,通过链霉菌的美登醇脱甲基化制备);和4,5-脱氧(美国第4,371,533号专利)(例如,通过美登醇的三氯化钛/lah还原制备)。
美登素类化合物上的许多位置可用作连接位置。例如,酯键可以通过使用常规偶联技术与羟基反应形成。在一些实施方案中,反应可以在具有羟基的c-3位置处发生,在用羟甲基修饰的c-14位置处发生,在用羟基修饰的c-15位置处发生,在具有羟基的c-20位置处发生。在一些实施方案中,连接在美登醇或美登醇类似物的c-3位置处形成。
美登素类化合物药物部分包括具有以下结构的那些:
其中波浪线表示美登素类化合物药物部分的硫原子与adc的接头的共价连接。每个r可以独立地为h或c1-c6烷基。将酰胺基团连接到硫原子上的亚烷基链可以是甲烷基、乙烷基或丙基,即m是1、2或3(美国第633,410号专利;美国第5,208,020号专利;chari等,cancerres.,第52卷,第127-131页,1992;liu等,proc.nall.acad.sci.usa,第93卷,第8618-8623页,1996)。
美登素类化合物药物部分的所有立体异构体都被考虑用于本发明的adc,即在手性碳上的r构型和s构型的任何组合(美国第7,276,497号专利;美国第6,913,748号专利;美国第6,441,163号专利;美国第633,410号专利(re39151);美国第5,208,020号专利;widdison等(2006)j.med.chem.49:4392-4408)。在一些实施方案中,美登素类化合物药物部分具有以下立体化学结构:
美登素类化合物药物部分的示例性实施方案包括但不限于具有以下结构的dm1、dm3和dm4:
其中波浪线表示药物的硫原子与抗体-药物缀合物的接头(l)的共价连接。
其中dm1通过bmpeo接头与抗体的硫醇基团连接的示例性抗体-药物缀合物具有以下结构和缩写:
其中ab是抗体;n为0、1或2;p为1至20。在一些实施方案中,p为1至10,p为1至7,p为1至5,或p为1至4。
含有美登素类化合物的免疫缀合物,其制备方法及其治疗用途公开于,例如,美国第5,208,020号和第5,416,064号专利;us2005/0276812a1;和欧洲专利ep0425235b1。还可参见liu等,proc.natl.acad.sci.usa,第93卷,第8618-8623页,1996;和chari等,cancerresearch,第52卷,第127-131页,1992。
在一些实施方案中,抗体-美登素类化合物缀合物可以通过将抗体与美登素类化合物分子化学连接而不显著降低抗体或美登素类化合物分子的生物活性来制备。参见,例如,美国第5,208,020号专利。在一些实施方案中,每个抗体分子缀合有平均3-4个美登素类化合物分子的adc已显示出增强靶细胞的细胞毒性的功效而不会不利地影响抗体的功能或溶解度。在某些情况下,与使用裸抗体相比,甚至预期一个分子的毒素/抗体会增强细胞毒性。
用于制备抗体-美登素类化合物缀合物的示例性连接基团包括,例如,本文所述的那些以及美国第5,208,020号专利;ep专利0425235b1;chari等,cancerresearch,第52卷,第127-131页,1992;us2005/0276812a1;和us2005/016993a1中公开的那些。
(2)奥利斯他汀和海兔毒素
药物部分包括海兔毒素、奥利斯他汀及其类似物和衍生物(美国第5,635,483号专利;美国第5,780,588号专利;美国第5,767,237号专利;美国第6,124,431号专利)。奥利斯他汀是海洋软体动物化合物海兔毒素-10的衍生物。尽管不意图受任何特定理论的束缚,但已显示海兔毒素和奥利斯他汀干扰微管动力学、gtp水解、核和细胞的分裂(woyke等,antimicrob.agentsandchemother.,第45卷,第3580-3584页,2001),并具有抗癌活性(美国第5,663,149号专利)和抗真菌活性(pettit等,antimicrob.agentschemother.,第42卷,第2961-2965页,1998)。海兔毒素/奥利斯他汀药物部分可以通过肽药物部分的n(氨基)末端或c(羧基)末端与抗体连接(wo02/088172;doronina等,naturebiotechnology,第21卷,第778-784页,2003;francisco等,blood,第102卷,第1458-1465页,2003)。
示例性的奥利斯他汀实施方案包括n-末端连接的单甲基奥利斯他汀药物部分de和df,其公开于美国第7,498,298号专利和美国第7,659,241号专利中:
其中de和df的波浪线表示与抗体或抗体-接头组分的共价连接位点,并且在每个位置独立地:
r2选自h和c1-c8烷基;
r3选自h、c1-c8烷基、c3-c8碳环、芳基、c1-c8烷基-芳基、c1-c8烷基-(c3-c8碳环)、c3-c8杂环和c1-c8烷基-(c3-c8杂环);
r4选自h、c1-c8烷基、c3-c8碳环、芳基、c1-c8烷基-芳基、c1-c8烷基-(c3-c8碳环)、c3-c8杂环和c1-c8烷基-(c3-c8杂环);
r5选自h和甲基;
或r4和r5共同形成碳环并具有下式-(crarb)n-,其中ra和rb独立地选自h、c1-c8烷基和c3-c8碳环,n选自2、3、4、5和6;
r6选自h和c1-c8烷基;
r7选自h、c1-c8烷基、c3-c8碳环、芳基、c1-c8烷基-芳基、c1-c8烷基-(c3-c8碳环)、c3-c8杂环和c1-c8烷基-(c3-c8杂环);
每个r8独立地选自h、oh、c1-c8烷基、c3-c8碳环和o-(c1-c8烷基);
r9选自h和c1-c8烷基;
r10选自芳基或c3-c8杂环;
z为o、s、nh或nr12,其中r12为c1-c8烷基;
r11选自h、c1-c20烷基、芳基、c3-c8杂环、-(r13o)m-r14、或-(r13o)m-ch(r15)2;
m为1-1000的整数;
r13为c2-c8烷基;
r14为h或c1-c8烷基;
每次出现的e独立地为h、cooh、-(ch2)n-n(r16)2、-(ch2)n-so3h或-(ch2)n-so3-c1-c8烷基;
每次出现的e独立地为h、c1-c8烷基或-(ch2)n-cooh;
r18选自-c(r8)2-c(r8)2-芳基、-c(r8)2-c(r8)2-(c3-c8杂环)和-c(r8)2-c(r8)2-(c3-c8碳环);
n为0至6的整数。
在一个实施方案中,r3、r4和r7独立地是异丙基或仲丁基,r5是-h或甲基。在一个示例性实施方案中,r3和r4各自为异丙基,r5为-h,r7为仲丁基。
在另一个实施方案中,r2和r6各自为甲基,r9为-h。
在另一个实施方案中,每次出现的r8是-och3。
在一个示例性实施方案中,r3和r4各自为异丙基,r2和r6各自为甲基,r5为-h,r7为仲丁基,每次出现的r8为-och3,r9为-h。
在一个实施方案中,z为-o-或-nh-。
在一个实施方案中,r10为芳基。
在一个示例性实施方案中,r10为-苯基。
在一个示例性实施方案中,当z为-o-时,r11为-h、甲基或叔丁基。
在一个实施方案中,当z为-nh时,r11为-ch(r15)2,其中r15为-(ch2)n-n(r16)2,r16为-c1-c8烷基或-(ch2)n-cooh。
在另一个实施方案中,当z为-nh时,r11为-ch(r15)2,其中r15为-(ch2)n-so3h。
式de的示例性奥利斯他汀实施方案为mmae,其中波浪线表示与抗体-药物缀合物的接头(l)的共价连接:
式de的示例性奥利斯他汀实施方案为mmaf,其中波浪线表示与抗体-药物缀合物的接头(l)的共价连接:
其他示例性实施方案包括在五肽奥利斯他汀药物部分的c-末端具有苯丙氨酸羧基修饰的单甲基缬氨酸化合物(wo2007/008848)和在五肽奥利斯他汀药物部分的c-末端具有苯丙氨酸侧链修饰的单甲基缬氨酸化合物(wo2007/008603)。
包含mmaf和各种接头组分的式i的adc的非限制性示例性实施方案还包括ab-mc-pab-mmaf和ab-pab-mmaf。包含不可通过蛋白水解切割的接头与抗体连接的mmaf的免疫缀合物已显示具有与包含可通过蛋白水解切割的接头与抗体连接的mmaf的免疫缀合物相当的活性(doronina等,bioconjugatechem.,第17卷,第114-124页,2006)。在一些这样的实施方案中,认为药物释放受细胞中抗体降解的影响。
通常,可以通过在两个或更多个氨基酸和/或肽片段之间形成肽键来制备基于肽的药物部分。这种肽键可以例如根据液相合成方法制备(参见例如e.
在一些实施方案中,式de(例如mmae)和de(例如mmaf)的奥利斯他汀/海兔毒素药物部分,和其药物-接头中间体及其衍生物,例如mc-mmaf、mc-mmae、mc-vc-pab-mmaf和mc-vc-pab-mmae,可以使用描述于以下中的方法制备:美国第7,498,298号专利;doronina等,bioconjugatechem.,第17卷,第114-124页,2006;和doronina等,nat.biotech.,第21卷,第778-784页,2003,然后与目的抗体缀合。
(3)卡奇霉素
在一些实施方案中,免疫缀合物包含与一个或多个卡奇霉素分子缀合的抗体。卡奇霉素家族抗生素及其类似物能够在亚皮摩尔浓度下产生双链dna断裂(hinman等,cancerresearch,第53卷,第3336-3342页,1993;lode等,cancerresearch,第58卷,第2925-2928页,1998)。卡奇霉素具有细胞内作用位点,但在某些情况下,不易穿过质膜。因此,在一些实施方案中,通过抗体介导的内化作用而进行的对这些药剂的细胞摄取可以极大地增强它们的细胞毒性作用。制备具有卡奇霉素药物部分的抗体-药物缀合物的非限制性示例性方法描述于,例如,美国第5,712,374号专利;美国第5,714,586号专利;美国第5,739,116号专利;和美国第5,767,285号专利中。
(4)吡咯并苯并二氮杂
在一些实施方案中,adc包含吡咯并苯并二氮杂
已经将pbd二聚体缀合至抗体,所得adc显示具有抗癌特性。pbd二聚体上的非限制性示例性连接位点包括五元吡咯环、pbd单元之间的系链和n10-c11亚胺基团(wo2009/016516;us2009/304710;us2010/047257;us2009/036431;us2011/0256157;wo2011/130598)。
adc的非限制性示例性pbd二聚体组分及其盐和溶剂化物如式a所示:
其中:
波浪线表示与接头的共价连接位点;
虚线表示c1和c2或c2和c3之间任选存在双键;
r2独立地选自h、oh、=o、=ch2、cn、r、or、=ch-rd、=c(rd)2、o-so2-r、co2r和cor,并且任选地进一步选自卤素或二卤素,其中rd独立地选自r、co2r、cor、cho、co2h和卤素;
r6和r9独立地选自h、r、oh、or、sh、sr、nh2、nhr、nrr’、no2、me3sn和卤素;
r7独立地选自h、r、oh、or、sh、sr、nh2、nhr、nrr’、no2、me3sn和卤素;
q独立地选自o、s和nh;
r11为h或r,或其中q为o、so3m,其中m为金属阳离子;
r和r’各自独立地选自任选取代的c1-8烷基、c1-12烷基、c3-8杂环基、c3-20杂环和c5-20芳基,并且任选地与基团nrr’、r及r’和它们所连接的氮原子一起形成任选取代的4-元杂环、5-元杂环、6-元杂环或7-元杂环;
r12、r16、r19和r17分别如r2、r6、r9和r7所定义;
r”是c3-12亚烷基,该链可以被一个或多个杂原子(例如,o、s、n(h),nme)和/或芳香环(例如,苯或吡啶)中断,其中环任选被取代;x和x’独立地选自o、s和n(h)。
在一些实施方案中,r和r’各自独立地选自任选取代的c1-12烷基、c3-20杂环和c5-20芳基,并且任选地与基团nrr’、r及r’和它们所连接的氮原子一起形成任选取代的4-元杂环、5-元杂环、6-元杂环或7-元杂环。在一些实施方案中,r9和r19为h。在一些实施方案中,r6和r16为h。
在一些实施方案中,r7和r17都为or7a,其中r7a是任选取代的c1-4烷基。在一些实施方案中,r7a为me。在一些实施方案中,r7a为ch2ph,其中ph为苯基。在一些实施方案中,x为o。在一些实施方案中,r11为h。在一些实施方案中,在每个单体单元中在c2和c3之间存在双键。
在一些实施方案中,r2和r12独立地选自h和r。在一些实施方案中,r2和r12独立地为r。在一些实施方案中,r2和r12独立地为任选取代的c5-20芳基或c5-7芳基或c8-10芳基。在一些实施方案中,r2和r12独立地是任选取代的苯基、噻吩基、萘基、吡啶基、喹啉基或异喹啉基。在一些实施方案中,r2和r12独立地选自=o、=ch2、=ch-rd和=c(rd)2。在一些实施方案中,r2和r12各自为=ch2。在一些实施方案中,r2和r12各自为h。在一些实施方案中,r2和r12各自为=o。在一些实施方案中,r2和r12各自为=cf2。在一些实施方案中,r2和/或r12独立地为=c(rd)2。在一些实施方案中,r2和/或r12独立地为=ch-rd。
在一些实施方案中,当r2和/或r12为=ch-rd时,每个基团可以独立地具有如下所示的任一构型:
在一些实施方案中,=ch-rd处于构型(i)中。在一些实施方案中,r”为c3亚烷基或c5亚烷基。
pbd二聚体-val-cit-pab-ab和pbd二聚体-phe-lys-pab-ab的接头是蛋白酶可切割的,而pbd二聚体-马来酰亚胺-缩醛的接头是酸不稳定的。
可以根据本领域已知的方法制备pbd二聚体和包含pbd二聚体的adc。参见,例如,wo2009/016516;us2009/304710;us2010/047257;us2009/036431;us2011/0256157;wo2011/130598。
(5)蒽环类药物
在一些实施方案中,adc可包含蒽环类药物。蒽环类药物是具有细胞毒活性的抗生素化合物。虽然不旨在受任何特定理论的束缚,但研究表明,蒽环类药物可通过多种不同机制杀死细胞,包括:1)将药物分子插入细胞dna中,从而抑制dna依赖性的核酸合成;2)由药物产生自由基然后与细胞大分子反应以对细胞造成损害,和/或3)药物分子与细胞膜的相互作用(参见,例如,c.peterson等,“transportandstorageofanthracyclineinexperimentalsystemsandhumanleukemia”inanthracyclineantibioticsincancertherapy;nrbachur,“freeradicaldamage”id.第97-102页)。由于其细胞毒性潜力,蒽环类药物已用于治疗多种癌症,例如白血病、乳腺癌、肺癌、卵巢腺癌和肉瘤(参见例如,p.h-wiernik,anthracycline:currentstatusandnewdevelopments,第11页)。
非限制性的示例性蒽环类药物包括多柔比星、表柔比星、伊达比星、道诺霉素、奈莫柔比星及其衍生物。已经制备并研究了道诺霉素和多柔比星的免疫缀合物和前药(kratz等,currentmed.chem.,第13卷,第477-523页,2006;jeffrey等,bioorganic&med.chem.letters,第16卷,第358-362页,1996;torgov等,bioconj.chem.,第16卷,第717-721页,2005;nagy等,proc.natl.acad.sci.usa,第97卷,第829-834页,2000;dubowchik等,bioorg.&med.chem.letters,第12卷,第1529-1532页,2002;king等,j.med.chem.,第45卷,第4336-4343页,2002;ep0328147;美国第6,630,579号专利)。抗体-药物缀合物br96-多柔比星与肿瘤相关抗原lewis-y发生特异性反应,并已在i期和ii期研究中进行评估(saleh等,j.clin.oncology,第18卷,第2282-2292页,2000;ajani等,cancerjour.,第6卷,第78-81页,2000;tolcher等,j.clin.oncology,第17卷,第478-484页,1999)。
pnu-159682是奈莫柔比星的强效代谢物(或衍生物)(quintieri等,clinicalcancerresearch,第11卷,第1608-1617页,2005)。奈莫柔比星是多柔比星的半合成类似物,在多柔比星的糖苷氨基上具有2-甲氧基吗啉基,并且已经进行了临床评估(grandi等,cancertreat.rev.第17卷,第133-138页,1990;ripamonti等,brit.j.cancer,第65卷,第703-707页,1992),包括用于肝细胞癌的ii/iii期试验(sun等,proceedingsoftheamericansocietyforclinicaloncology,第22卷,abs1448,2003;quintieri,proceedingsoftheamericanassociationofcancerresearch,第44卷:第1版,abs4649,2003;pacciarini等,jour.clin.oncology,第24卷,第14116页,2006)。
蒽环类药物,包括pnu-159682,可以通过几个连接位点和多种接头(包括本文所述的接头)与抗体缀合(us2011/0076287;wo2009/099741;us2010/0034837;wo2010/009124)。
pnu-159682马来酰亚胺缩醛-ab的接头是酸不稳定的,而pnu-159682-val-cit-pab-ab的接头、pnu-159682-val-cit-pab-间隔物-ab的接头和pnu-159682-val-cit-pab-间隔物(r1r2)-ab的接头是蛋白酶可切割的。
(6)其他药物部分
药物部分还包括格尔德霉素(mandler等,j.nat.cancerinst.,第92卷,第1573-1581页,2000;mandler等,bioorganic&med.chem.letters,第10卷,第1025-1028页,2000;mandler等,bioconjugatechem.,第13卷,第786-791页,2002);和酶活性毒素及其片段,包括但不限于白喉毒素a链,白喉毒素的非结合活性片段,外毒素a链(来自铜绿假单胞菌)、蓖麻毒蛋白a链、相思豆毒蛋白a链、蒴莲根毒蛋白a链、α-帚曲霉素、油桐蛋白、香石竹毒蛋白、美洲商陆蛋白(papi、papii和pap-s)、苦瓜抑制剂、麻疯树毒蛋白、巴豆毒蛋白、肥皂草抑制剂、白树毒蛋白、丝林霉素、局限曲菌素、酚霉素、依诺霉素和单端孢霉烯族化合物。参见,例如,wo93/21232。
药物部分还包括具有核溶解活性的化合物(例如,核糖核酸酶或dna核酸内切酶)。
在某些实施方案中,免疫缀合物可包含高放射性原子。有多种放射性同位素可用于生产放射性缀合抗体。实例包括at211、i131、i125、y90、re186、re188、sm153、bi212、p32、pb212和lu的放射性同位素。在一些实施方案中,当免疫缀合物用于检测时,其可包含用于闪烁扫描研究的放射性原子,例如tc99或i123,或用于核磁共振(nmr)成像(也称为磁共振成像,mri)的自旋标记,例如锆-89、碘-123、碘-131、铟-111、氟-19、碳-13、氮-15、氧-17、钆、锰或铁。锆-89可以与各种金属螯合剂络合并与抗体缀合,用于例如pet成像(wo2011/056983)。
可以以已知方式将放射性标记或其他标记掺入免疫缀合物中。例如,可以使用合适的氨基酸前体生物合成肽或化学合成肽,所述氨基酸前体包括例如取代一个或多个氢的一个或多个氟-19原子。在一些实施方案中,诸如tc99、i123、re186、re188和in111的标记可以通过抗体中的半胱氨酸残基连接。在一些实施方案中,钇-90可以通过抗体的赖氨酸残基连接。在一些实施方案中,iodogen方法(fraker等,biochem.biophys.res.commun.,第80卷,第49-57页,1978)可用于掺入碘-123。“monoclonalantibodiesinimmunoscintigraphy”(chatal,crcpress1989)描述了某些其他方法。
在某些实施方案中,免疫缀合物可包含与前药活化酶缀合的抗体。在一些这样的实施方案中,前药活化酶将前药(例如,肽基化学治疗剂,参见wo81/01145)转化为活性药物,例如抗癌药物。在一些实施方案中,此类免疫缀合物可用于抗体依赖性的由酶介导的前药治疗(“adept”)。可以与抗体缀合的酶包括但不限于碱性磷酸酶,其可用于将含磷酸酯基的前药转化为游离药物;芳基硫酸酯酶,可用于将含硫酸酯基的前药转化为游离药物;胞嘧啶脱氨酶,可用于将无毒的5-氟胞嘧啶转化为抗癌药物5-氟尿嘧啶;蛋白酶,例如沙雷菌(serratia)蛋白酶、嗜热菌蛋白酶、枯草杆菌蛋白酶、羧肽酶和组织蛋白酶(例如组织蛋白酶b和l),可用于将含有肽的前药转化为游离药物;d-丙氨酰羧肽酶,可用于转化含有d-氨基酸取代基的前药;碳水化合物裂解酶,例如β-半乳糖苷酶和神经氨酸酶,可用于将糖基化前药转化为游离药物;β-内酰胺酶,可用于将用β-内酰胺衍生的药物转化为游离药物;和青霉素酰胺酶,例如青霉素v酰胺酶和青霉素g酰胺酶,它们可用于将分别用苯氧基乙酰基或苯乙酰基在它们的氨基氮处衍生的药物转化成游离药物。在一些实施方案中,酶可以通过本领域熟知的重组dna技术与抗体共价结合。参见,例如,neuberger等,nature,第312卷,第604-608页,1984。
c)载药量
载药量由p表示,p是式i分子中每抗体的药物部分平均数。载药量范围可以为每个抗体1至20个药物部分(d)。式i的adc包括与1至20个药物部分缀合的抗体的集合。在通过缀合反应制备adc中每个抗体使用的药物部分的平均数可以通过常规手段例如质谱、elisa测定法和hplc进行表征。还可以确定以p表示的adc的定量分布。在一些情况下,可以通过诸如反相hplc或电泳的方法从具有其他载药量的adc中分离、纯化和表征其中p为特定值的均质adc。
对于一些抗体-药物缀合物,p可能受抗体上连接位点的数量的限制。例如,如以上某些示例性实施方案中所述,在连接位点是半胱氨酸硫醇基的情况下,抗体可以仅具有一个或几个半胱氨酸硫醇基团,或者可以仅具有一个或几个具有足够反应性的硫醇基团,接头可以通过所述硫醇基团连接。在某些实施方案中,较高的载药量,例p>5,可能导致某些抗体-药物缀合物的聚集、不溶性、毒性、或细胞渗透性丧失。在某些实施方案中,adc的平均载药量范围为1至约8;约2至约6;或者约3至约5。实际上,已经表明,对于某些adc,每个抗体的药物部分的最佳比例可以小于8,可以为约2至约5(美国第7,498,298号专利)。
在某些实施方案中,在缀合反应期间,少于理论最大值的药物部分与抗体缀合。如下所述,抗体可含有例如不与药物-接头中间体或接头试剂反应的赖氨酸残基。通常,抗体不含有多个可与药物部分连接的游离的和反应性的半胱氨酸硫醇基团;实际上,抗体中的大多数半胱氨酸硫醇残基以二硫键存在。在某些实施方案中,可以在部分还原条件或全部还原条件下用还原剂例如二硫苏糖醇(dtt)或三羰基乙基膦(tcep)还原抗体,以产生反应性半胱氨酸硫醇基团。在某些实施方案中,使抗体经历变性条件以暴露反应性亲核基团,例如赖氨酸或半胱氨酸。
可以以不同方式控制adc的载药量(药物/抗体比),例如,通过:(i)限制相对于抗体摩尔过量的药物-接头中间体或接头试剂,(ii)限制缀合反应时间或温度,和(iii)用于半胱氨酸硫醇基修饰的部分还原条件或限制性还原条件。
应当理解,当多于一个亲核基团与药物-接头中间体或接头试剂反应时,所得产物是adc的混合物,其具有与抗体连接的一个或多个药物部分的分布。可以通过双重elisa抗体测定法(其对抗体是特异性的且对药物是特异性的)计算混合物中每个抗体的药物平均数。可以通过质谱法在混合物中鉴定单个adc,并通过hplc分离单个adc,例如通过疏水性相互作用色谱(参见,例如,mcdonagh等,prot.engr.design&selection,第19卷,第299-307页,2006;hamblett等,clin.cancerres.,第10卷,第7063-7070页,2004)分离单个adc。在某些实施方案中,可以通过电泳或色谱法从缀合物混合物中分离具有单一负载值的均质adc。
d)制备免疫缀合物的一些方法
可以通过使用本领域技术人员已知的有机化学反应、条件和试剂的几种途径制备如式i所示adc的免疫缀合物,包括:(1)抗体的亲核基团与二价接头试剂反应以通过共价键形成ab-l,然后与药物部分d反应;(2)药物部分的亲核基团与二价接头试剂反应,以通过共价键形成d-l,然后与抗体的亲核基团反应。通过后一种途径制备式i的adc的示例性方法描述于美国第7,498,298号专利中。
抗体上的亲核基团包括但不限于:(i)n-末端氨基,(ii)侧链氨基,例如,赖氨酸,(iii)侧链硫醇基团,例如,半胱氨酸,和(iv)糖基化抗体中糖的羟基或氨基。氨基、硫醇基和羟基是亲核的,能够与接头部分上的亲电子基团反应而形成共价键,而接头试剂包括:(i)活性酯类,例如nhs酯、hobt酯、卤代甲酸酯和酸性卤化物;(ii)烷基和苄基卤化物,例如卤代乙酰胺;(iii)醛类、酮类、羧基和马来酰亚胺基团。某些抗体具有可还原的链间二硫键,即半胱氨酸桥。通过用还原剂例如dtt(二硫苏糖醇)或三羰基乙基膦(tcep)处理使得抗体完全还原或部分还原,从而可以使抗体具有与接头试剂缀合的反应活性。因此,每个半胱氨酸桥理论上将形成两个反应性硫醇基亲核体。可通过赖氨酸残基的修饰将额外的亲核基团引入抗体中,例如通过使赖氨酸残基与2-亚氨基硫烷(traut氏试剂)反应,使氨基转变为硫醇基。还可以通过引入一个、两个、三个、四个或更多个半胱氨酸残基(例如,通过制备包含一个或多个非天然半胱氨酸氨基酸残基的抗体变体)将反应性硫醇基团引入抗体中。
本发明的抗体-药物缀合物还可以通过抗体上的亲电子基团(例如醛基或酮羰基)与接头试剂或药物上的亲核基团之间的反应来产生。接头试剂上有用的亲核基团包括但不限于酰肼、肟、氨基、肼、缩氨基硫脲、羧酸肼和芳基酰肼。在一个实施方案中,修饰抗体以引入能够与接头试剂或药物上的亲核取代基反应的亲电子部分。在另一个实施方案中,糖基化抗体的糖可以被例如高碘酸盐氧化剂氧化,形成可以与接头试剂或药物部分的氨基反应的醛基或酮基。得到的亚胺席夫碱基可以形成稳定的键,或者可以被例如硼氢化物试剂还原,形成稳定的胺键。在一个实施方案中,糖基化抗体的碳水化合物部分与半乳糖氧化酶或偏高碘酸钠的反应可在抗体中产生羰基(醛和酮)基团,其可与药物上的适当基团反应(hermanson,bioconjugatetechniques)。在另一个实施方案中,含有n-末端丝氨酸或苏氨酸残基的抗体可以与偏高碘酸钠反应,从而导致在第一个氨基酸的位置产生醛(geoghegan&stroh,bioconjugatechem.,第3卷,第138-146页,1992;美国第5,362,852号专利)。这种醛可以与药物部分或接头亲核体反应。
药物部分上的示例性亲核基团包括但不限于:氨基、硫醇基团、羟基、酰肼基团、肟基团、肼基团、缩氨基硫脲基团、羧酸肼基团和芳基酰肼基团,其能够与接头部分上的亲电子基团反应形成共价键,而接头试剂包括:(i)活性酯类,例如nhs酯、hobt酯、卤代甲酸酯和酸性卤化物;(ii)烷基和苄基卤化物,例如卤代乙酰胺;(iii)醛类、酮类、羧基和马来酰亚胺基团。
可用于制备adc的非限制性的示例性交联剂试剂在本文的标题为“示例性接头”的部分中描述。使用此类交联剂试剂连接两个部分(包括蛋白部分和化学部分)的方法,在本领域中是已知的。在一些实施方案中,可以例如通过重组技术或肽合成制备包含抗体和细胞毒性剂的融合蛋白。重组dna分子可包含编码抗体和编码缀合物的细胞毒性部分的区域,它们彼此相邻或由编码接头肽的区域隔开,所述接头肽不破坏缀合物的所需特性。
在另一个实施方案中,抗体可以与“受体”(例如链霉亲和素)缀合,用于肿瘤预靶向,其中将抗体-受体缀合物施用给患者,接着使用清除剂从循环中除去未结合的缀合物,然后施用与细胞毒性剂(例如药物或放射性核苷酸)缀合的“配体”(例如,抗生物素蛋白)。
c.用于诊断和检测的方法和组合物
在某些实施方案中,本文提供的任何抗axl抗体或抗体片段可用于检测生物样品中axl的存在。本文中使用的术语“检测”包括定量检测或定性检测。在某些实施方案中,生物样品包含细胞或组织,例如乳腺细胞或组织、胰腺细胞或组织、食道细胞或组织、肺细胞或组织和/或脑细胞或组织。
本发明的另一方面涉及本发明的抗axl抗体,其用于诊断和/或监测癌症或其他疾病,在所述癌症或其他疾病中,axl水平在身体的至少一个部位与正常生理水平相比增加或减少。
在一个优选的实施方案中,本发明的抗体或抗体片段可以用可检测的分子或物质标记,例如用萦光分子、放射性分子或如上所述的本领域已知的任何其他标记物标记。例如,可以用放射性分子标记本发明的抗体。例如,合适的放射性分子包括但不限于用于闪烁扫描研究的放射性原子,例如123i、124i、111in、186re和188re。本发明的抗体或抗体片段也可以用用于核磁共振(nmr)成像的自旋标记物标记,例如用碘-123、碘-131、铟-ill、氟-19、碳-13、氮-15、氧-17、钆、锰或铁标记。施用抗体后,检测放射性标记的抗体在患者体内的分布。可以使用任何合适的已知方法。一些非限制性实例包括计算机断层摄影(ct)、位置发射断层摄影(pet)、磁共振成像(mri)、萦光、化学发光和超声检查。
本发明的抗体或抗体片段可用于与axl过表达相关的癌症和疾病的诊断和分期。与axl过表达相关的癌症可包括鳞状细胞癌、小细胞肺癌、非小细胞肺癌、胃癌、胰腺癌、胶质细胞瘤(例如胶质母细胞瘤和多发性神经纤维瘤)、宫颈癌、卵巢癌、肝癌、膀胱癌、肝细胞瘤、乳腺癌、结肠癌、黑色素瘤、结肠直肠癌、子宫内膜癌、唾液腺癌、肾癌、肾肿瘤、前列腺癌、外阴癌、甲状腺癌、肝肿瘤、肉瘤、血液癌(白血病)、星形细胞瘤、和各种类型的头颈癌或其他axl表达或过度表达的过度增生性疾病。
本发明的抗体或抗体片段可用于诊断除癌症以外的axl表达增加或减少的疾病。可溶性axl形式或细胞axl形式均可用于此类诊断。通常,此类诊断方法涉及使用从患者获得的生物样品。本文使用的术语“生物样品”包括从可以用于诊断或监测测定的个体获得的多种样品类型。生物样品包括但不限于生物来源的血液和其他液体样品,和实体组织样品,例如,活组织检查样品或组织培养物或由其衍生的细胞及其后代。例如,生物样品包括从怀疑患有与axl过表达相关的癌症的个体收集的组织样品中获得的细胞,在优选的实施方案中,来自患有神经胶质瘤、胃癌、肺癌、胰腺癌、乳腺癌、前列腺癌、肾癌、肝癌和子宫内膜癌的个体。生物样品包括临床样品、培养细胞、细胞上清液、细胞裂解液、血清、血浆、生物液和组织样品。
在一个具体实施方案中,本发明是通过使用本发明的抗体检测来自个体的细胞上的axl来诊断该个体中与axl过表达相关的癌症的方法。特别地,所述方法可包括以下步骤:
(a)在适合抗体或抗体片段与生物样品中表达axl的细胞形成复合物的条件下,使个体的生物样品与本发明的抗体或抗体片段接触;
(b)检测和/或定量化所述复合物,由此所述复合物的检测指示与axl过表达相关的癌症。
为了监测癌症的发展,可以在不同时间重复根据本发明的方法,以确定抗体与样品的结合是增加还是减少,由此可以确定癌症是否已经发展、消退或稳定。
在一个具体实施方案中,本发明是诊断与axl的表达或过表达或axl的可溶形式的减少或增加相关的疾病的方法。此类疾病的实例可包括人体免疫紊乱、血栓性疾病(血栓形成和动脉粥样硬化血栓形成)和心血管疾病。
在一个实施方案中,提供了用于诊断或检测方法中的抗axl抗体或抗体片段。在另一方面,提供了一种检测生物样品中axl的存在的方法。在另一方面,提供了一种量化生物样品中axl的量的方法。在某些实施方案中,该方法包括在允许抗axl抗体或抗体片段与axl结合的条件下使生物样品与本文所述的抗axl抗体或抗体片段接触,并检测在抗ax1抗体或抗体片段和axl之间是否形成复合物。这种方法可以在体外或体内进行。在一个实施方案中,抗axl抗体或抗体片段用于选择适合治疗的个体。在一些实施方案中,治疗包括向个体施用抗axl抗体或抗体片段。
在某些实施方案中,提供了标记的抗axl抗体或抗体片段。标记物包括但不限于可直接检测的标记物或部分(例如萦光标记物、发色标记物、电子致密标记物、化学发光标记物和放射性标记物),以及可通过例如酶促反应或分子相互作用而间接检测的诸如酶或配体的部分。示例性标记物包括但不限于放射性同位素(32p、14c、125i、3h和131i)、萦光团(例如稀土螯合物或萦光素及其衍生物、罗丹明及其衍生物、丹酰基、伞形酮)、半乳糖和氯氨素乙烯转化酶(luceriferases,例如萤火虫萦光素酶和细菌萦光素酶(美国第4,737,456号专利))、萦光素、2,3-二氢酞嗪二酮、辣根过氧化物酶(hrp)、碱性磷酸酶、β-半乳糖苷酶、葡糖淀粉酶、溶菌酶、糖氧化酶(例如葡萄糖氧化酶、半乳糖氧化酶和葡萄糖-6-磷酸脱氢酶)、杂环氧化酶(例如尿酸酶和黄嘌呤氧化酶,与利用过氧化氢氧化染料前体的酶(例如hrp、乳过氧化物酶或微过氧化物酶)相偶联)、生物素/抗生物素蛋白、自旋标记物、噬菌体标记物、稳定自由基等。
d.药物制剂
抗axl抗体或抗体片段具有细胞杀伤活性。该细胞杀伤活性适用范围扩展到多种不同类型的细胞系。此外,这些抗体或抗体片段一旦与细胞毒性剂缀合,就可以减小肿瘤尺寸并且可以表现出降低的毒性。参见本申请的实施例3和6-9。因此,抗axl抗体、其片段或免疫缀合物可用于治疗与axl表达相关的增生性疾病。抗体、抗体片段或免疫缀合物可以单独使用或与任何合适的药剂或其他常规治疗组合使用。
抗axl抗体或抗体片段可用于治疗与axl和/或gas6表达,过表达或活化相关的过度增生性疾病。除了对axl表达的要求之外,对可以治疗的癌症或组织的类型没有特别限制。实例包括鳞状细胞癌、小细胞肺癌、非小细胞肺癌、胃癌、胰腺癌、胶质细胞瘤(例如胶质母细胞瘤和多发性神经纤维瘤)、宫颈癌、卵巢癌、肝癌、膀胱癌、肝细胞瘤、乳腺癌、结肠癌、黑色素瘤、结肠直肠癌、子宫内膜癌、唾液腺癌、肾癌、肾肿瘤、前列腺癌、外阴癌、甲状腺癌、肝肿瘤、肉瘤、血液癌(白血病)、星形细胞瘤、和各种类型的头颈癌。更优选的癌症是神经胶质瘤、胃癌、肺癌、胰腺癌、乳腺癌、前列腺癌、肾癌、肝癌和子宫内膜癌。
抗axl抗体或抗体片段是先天免疫应答的潜在激活剂,因此可用于治疗人免疫疾病,例如治疗败血症。本发明的抗axl抗体或抗体片段也可用作免疫的佐剂(例如疫苗)和抗感染剂(例如抗细菌剂、抗病毒剂和抗寄生虫剂)。
抗axl抗体或抗体片段可用于防范、预防或治疗血栓性疾病(例如静脉和动脉血栓形成和动脉粥样硬化血栓形成)。抗axl抗体或抗体片段也可用于防范、预防或治疗心血管疾病以及预防或抑制病毒(例如拉沙病毒和埃博拉病毒)的入侵以及治疗病毒感染。
在本文所述的治疗方法的每个实施方案中,抗axl单克隆抗体、抗体片段或抗axl单克隆免疫缀合物可以以与管理所要治疗的疾病或病症相关的常规方法一致的方式递送。根据本文的公开内容,在足以预防或治疗疾病或病症的条件下,将有效量的抗体、抗体片段或免疫缀合物向需要这种治疗的个体施用一段时间。因此,本发明的一个方面涉及治疗与axl表达相关的疾病的方法,该方法包括向有此需要的个体施用治疗有效量的本发明的抗体、抗体片段或免疫缀合物。
对于施用而言,可以将抗axl单克隆抗体、抗体片段或免疫缀合物配制成药物组合物。包含抗axl单克隆抗体、抗体片段或抗体-药物缀合物的药物组合物可根据制备药物组合物的已知方法配制。在这些方法中,治疗分子通常与含有药学上可接受的载体的混合物、溶液或组合物组合。
药学上可接受的载体是接受治疗的患者可以耐受的材料。无菌磷酸盐缓冲盐水是药学上可接受的载体的一个实例。其他合适的药学上可接受的载体是本领域技术人员所熟知的(参见,例如,gennaro(编辑),remington'spharmaceuticalsciences(mackpublishingcompany,第19版,1995))。制剂可进一步包括防止小瓶表面上的蛋白损失的一种或多种赋形剂、防腐剂、增溶剂、缓冲剂、白蛋白等。
药物组合物的形式,施用途径,剂量和方案自然取决于待治疗的病症、疾病的严重程度、患者的年龄、体重和性别等。本领域技术人员可以考虑这些因素配以制合适的药物组合物。本发明的药物组合物可以配制成用于外用、口服、肠胃外施用、鼻内施用、静脉内施用、肌肉内施用、皮下施用或眼内施用等。
优选地,药物组合物含有对于能够注射的制剂而言在药学上可接受的载体。这些可以具体地是等渗的、无菌的、盐水溶液(磷酸一钠或磷酸二钠、氯化钠、氯化钾、氯化钙或氯化镁等或这些盐的混合物),或干燥的、特别是冻干的组合物,根据情况,例如在添加无菌水或生理盐水后,所述组合物允许形成注射溶液。
在一些实施方案中,存在张度剂,有时称为“稳定剂”以调节或维持组合物中液体的张力。当与大的带电生物分子(例如蛋白和抗体)一起使用时,它们通常被称为“稳定剂”,因为它们可以与氨基酸侧链的带电基团相互作用,从而减少分子间相互作用和分子内相互作用的可能性。张度剂可以以药物组合物的0.1-25重量%,优选1-5重量%的任何量存在。优选的张度剂包括多元糖醇,优选三元糖醇或更高级糖醇,例如甘油、赤藓糖醇、阿拉伯糖醇、木糖醇、山梨糖醇和甘露糖醇。
其他赋形剂包括可用作以下中的一种或多种的试剂:(1)填充剂,(2)溶解度增强剂,(3)稳定剂和(4)防止变性或粘附到容器壁上的试剂。这些赋形剂可包括:多元糖醇类(如以上所列);氨基酸类,例如丙氨酸、甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸、赖氨酸、鸟氨酸、亮氨酸、2-苯丙氨酸、谷氨酸、苏氨酸等;有机糖类或糖醇类,例如蔗糖、乳糖、乳糖醇、海藻糖、水苏糖、甘露糖、山梨糖、木糖、核糖、核糖醇、肌糖(myoinisitose)、肌醇(myoinisitol)、半乳糖、半乳糖醇、甘油、环醇(例如肌醇)、聚乙二醇;含硫还原剂,例如尿素、谷胱甘肽、硫辛酸、硫代乙醇酸钠、硫代甘油、α-硫代甘油和硫代硫酸钠;低分子量蛋白,例如人血清白蛋白、牛血清白蛋白、明胶或其他免疫球蛋白;亲水性聚合物,例如聚乙烯吡咯烷酮;单糖类(例如,木糖、甘露糖、果糖、葡萄糖);二糖类(例如乳糖、麦芽糖、蔗糖);三糖类例如棉子糖;和多糖类例如糊精或葡聚糖。
可以使用非离子表面活性剂或清洁剂(也称为“润湿剂”)来帮助溶解治疗剂以及保护治疗蛋白免受由搅动诱导的聚集,这也允许制剂暴露于剪切表面应力而不引起活性治疗蛋白或抗体的变性。非离子表面活性剂可以以约0.05mg/ml至约1.0mg/ml,优选约0.07mg/ml至约0.2mg/ml的浓度范围存在。
合适的非离子表面活性剂包括聚山梨醇酯类(20、40、60、65、80等)、泊洛沙姆(polyoxamers184、188等)、
用于施用的剂量可以根据各种参数进行调整,特别是根据施用方式、相关病理或所需的治疗持续时间进行调整。为了制备药物组合物,可以将有效量的抗体或抗体片段溶解或分散在药学上可接受的载体或水性介质中。
适于注射使用的药物剂型包括无菌水溶液或分散液;包含芝麻油、花生油或丙二醇水溶液的制剂;和用于临时制备无菌可注射溶液或分散液的无菌粉末。在所有情况下,该剂型必须是无菌的且流动性必须达到易于注射的程度。它在制造和储存条件下必须是稳定的,并且必须免受微生物(例如细菌和真菌)的污染。
可以在适当地与表面活性剂混合的水中制备作为游离碱或药理学上可接受的盐的活性化合物的溶液。分散液也可以在甘油、液体聚乙二醇及其混合物和油中制备。在通常的储存和使用条件下,这些制剂含有防腐剂以防止微生物的生长。
可以将抗体或抗体片段配制成中性形式或盐形式的组合物。药学上可接受的盐包括酸加成盐(与蛋白的游离氨基形成),其用无机酸(例如盐酸或磷酸)形成,或用有机酸(例如乙酸、草酸、酒石酸、扁桃酸等)形成。与游离羧基形成的盐也可以衍生自无机碱(例如氢氧化钠、氢氧化钾、氢氧化铵、氢氧化钙或氢氧化铁),和有机碱(例如异丙胺、三甲胺、组氨酸、普鲁卡因等)。
载体也可以是溶剂或分散介质,其包括例如水、乙醇、多元醇(例如甘油、丙二醇和液体聚乙二醇等)、它们合适的混合物和植物油。通过以下方式可以保持适当的流动性,例如,通过使用涂层(例如卵磷脂),通过在分散的情况下维持所需的粒度和通过使用表面活性剂。可以通过各种抗细菌剂和抗真菌剂,例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸、硫柳汞等来防止微生物的作用。在许多情况下,优选包括等渗剂,例如糖类或氯化钠。通过在组合物中使用延迟吸收剂,例如单硬脂酸铝和明胶,可以实现可注射组合物的延长吸收。
通过将活性化合物以所需的量掺入具有上面列举的一种或多种其他成分的适当溶剂中,(如果需要)然后进行过滤灭菌来制备无菌可注射溶液。通常,通过将各种经灭菌的活性成分掺入无菌载体中来制备分散液,所述无菌载体含有基础分散介质和来自上面列举的那些中的所需其他成分。在用于制备无菌可注射溶液的无菌粉末的情况下,优选的制备方法是真空干燥和冷冻干燥技术,其由先前经无菌过滤的溶液产生活性成分和任何其他所需成分的粉末。
还考虑制备更浓缩的或高浓度的直接注射溶液,其中设想使用二甲基亚砜(dmso)作为溶剂导致极快的渗透,将高浓度的活性剂递送至小范围的肿瘤区域。
在配制后,溶液将以与剂量配方相容的方式并以治疗有效的量施用。制剂易于以多种剂型施用,例如以上述可注射溶液的剂型施用,但也可使用药物释放胶囊等。
例如,对于以水性溶液形式进行的肠胃外施用,水性溶液在必要时应该适当地进行缓冲,应该用足够的盐水或葡萄糖使液体稀释剂等渗。这些特定的水性溶液特别适用于静脉内施用,肌肉内施用,皮下施用和腹膜内施用。就此而言,根据本发明,可以使用的无菌水性介质对于本领域技术人员而言是已知的。例如,可以将一个剂量溶解在1ml等渗nacl溶液中,将其添加到1000ml的皮下注射液中或者在建议的输注部位注射(参见例如,“remington'spharmaceuticalsciences”第15版,第1035-1038页和第1570-1580页)。取决于所治疗的个体的状况,必然会出现剂量的一些变化。在任何情况下,负责施用的人员将确定对于单个个体的适当剂量。
可以在治疗混合物中配制抗体或抗体片段,以每剂约0.0001至10.0毫克,或约0.001至5毫克,或约0.001至1毫克,或约0.001至0.1毫克,或约0.1至1.0毫克或甚至每剂约10毫克递送。也可以以选定的时间间隔施用多个剂量。
除了配制用于肠胃外施用(例如静脉内注射或肌内注射)的化合物以外,其他药学上可接受的剂型包括,例如,用于口服施用的片剂或其他固体;定时释放胶囊;以及目前使用的任何其他剂型。
在某些实施方案中,考虑使用脂质体和/或纳米颗粒将抗体或抗体片段引入宿主细胞中。脂质体和/或纳米颗粒的形成和使用是本领域技术人员已知的。
纳米胶囊通常可以以稳定且可再现的方式捕获化合物。为了避免由于细胞内聚合物过载造成的副作用,通常使用能够在体内降解的聚合物设计这种超细颗粒(尺寸为约0.1μm)。本发明中使用符合这些要求的可生物降解的聚氰基丙烯酸烷基酯纳米颗粒,且这种颗粒易于制备。
由分散在水性介质中并自发形成多层包围式双层囊泡(也称作多层脂囊(mlv))的磷脂可形成脂质体。mlv的直径通常是25nm-4μm。超声处理mlv可形成直径为
通过将具有所需纯度的抗体或抗体片段与一种或多种任选的药学上可接受的载体混合来制备含有如本文所述的抗axl抗体或抗体片段的药物制剂(remington'spharmaceuticalsciences,第16版,osol,a.编辑(1980)),其剂型为冻干制剂或水性溶液。药学上可接受的载体通常在所用剂量和浓度下对接受者无毒,其包括但不限于:缓冲剂,例如磷酸盐、柠檬酸盐和其他有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸;防腐剂(例如十八烷基二甲基苄基氯化铵;氯化六甲双铵;苯扎氯铵;苄索氯铵;苯酚、丁基醇或苄基醇;对羟基苯甲酸烷基酯,例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇;和间甲酚);低分子量(少于约10个残基)的多肽;蛋白类,例如血清白蛋白、明胶或免疫球蛋白;亲水性聚合物,例如聚乙烯吡咯烷酮;氨基酸类,例如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或赖氨酸;单糖类、二糖类和其他碳水化合物类,包括葡萄糖、甘露糖或糊精;螯合剂例如edta;糖类,例如蔗糖、甘露醇、海藻糖或山梨糖醇;成盐反离子例如钠;金属配合物(例如锌-蛋白配合物);和/或非离子表面活性剂,例如聚乙二醇(peg)。
本文的示例性的药学上可接受的载体还包括间质药物分散剂,例如可溶性中性-活性透明质酸酶糖蛋白(shasegp),例如人可溶性ph-20透明质酸酶糖蛋白,例如rhuph20(
示例性的冻干抗体制剂描述于美国第6,267,958号专利中。水性抗体制剂包括描述于美国第6,171,586号专利和wo2006/044908中的那些,后一种制剂包括组氨酸-乙酸盐缓冲液。
本文的制剂还可含有一种以上的治疗具体适应症所必需的活性成分。优选地,具有互补活性且彼此不产生不利影响的成分可以组合成单一制剂。例如,除了本发明的抗axl抗体、抗体片段或免疫缀合物之外,可能需要提供egfr拮抗剂(例如厄洛替尼)、抗血管生成剂(例如可以是抗vegf抗体的vegf拮抗剂)或化学治疗剂(例如紫杉烷或铂剂)。这些活性成分适当地以对预期目的有效的量组合存在。
可以将活性成分包封在例如通过凝聚技术或通过界面聚合制备的微胶囊中。例如,可以使用分别在胶体药物递送系统(例如,脂质体、白蛋白微球、微乳液、纳米颗粒和纳米胶囊)或粗乳液中的羟甲基纤维素或明胶-微胶囊和聚-(甲基丙烯酸甲酯)微胶囊。这些技术公开于remington'spharmaceuticalsciences第16版,osol,a.编辑(1980)。
可以制备持续释放制剂。持续释放制剂的合适实例包括含有抗体或抗体片段的固体疏水聚合物的半透性基质,该基质可以成型制品(例如,膜或微胶囊)的形式存在。
用于体内施用的制剂通常是无菌的。例如,通过无菌过滤膜过滤可以较容易地实现无菌。
e.治疗方法和组合物
本文提供的任何抗axl抗体或抗体片段可用于治疗方法。在一个方面,提供了用作药物的抗axl抗体或抗体片段。在其他方面,提供了用于治疗癌症的抗axl抗体或抗体片段,所述癌症包括例如,乳腺癌、非小细胞肺癌、胰腺癌、脑癌、胰腺癌、脑癌、肾癌、卵巢癌、胃癌、白血病、子宫内膜癌、结肠癌、前列腺癌、甲状腺癌、肝癌、骨肉瘤和/或黑素瘤。在某些实施方案中,提供了用于治疗方法的抗axl抗体或抗体片段。在某些实施方案中,本发明提供了用于治疗患有癌症的个体的方法中的抗axl抗体或抗体片段,所述方法包括向个体施用有效量的抗axl抗体或抗体片段。在某些实施方案中,本发明提供了抗axl抗体或抗体片段,其用于治疗患有免疫疾病(例如,自身免疫性疾病)、心血管疾病(例如,动脉粥样硬化、高血压、血栓形成)、传染病(例如,埃博拉病毒、马尔堡病毒)或糖尿病的个体的方法,所述方法包括向个体施用有效量的抗axl抗体或抗体片段。在一个这样的实施方案中,例如,如下所述,该方法还包括向个体施用有效量的至少一种其他治疗剂。在进一步的实施方案中,本发明提供抗axl抗体或抗体片段,其用于抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统(例如肿瘤内脉管系统或肿瘤相关脉管系统)和/或抑制肿瘤基质功能。
在某些实施方案中,本发明提供抗axl抗体或抗体片段,其用于在个体中抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统(例如,肿瘤内脉管系统或肿瘤相关脉管系统),和/或抑制肿瘤基质功能的方法中,所述方法包括向个体施用有效量的抗axl抗体或抗体片段以抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统发育(例如,抑制肿瘤内脉管系统或肿瘤相关脉管系统发育),和/或抑制肿瘤基质功能。根据任何上述实施方案的“个体”优选是人。
在另一方面,本发明提供抗axl抗体或抗体片段在制造或制备药物中的用途。在一个实施方案中,所述药物用于治疗癌症(在一些实施方案中,包括乳腺癌、非小细胞肺癌、胰腺癌、脑癌、胰腺癌、脑癌、肾癌、卵巢癌、胃癌、白血病、子宫内膜癌、结肠癌、前列腺癌、甲状腺癌、肝癌、骨肉瘤和/或黑色素瘤)。在另一实施方案中,所述药物用于治疗癌症的方法中,所述方法包括向患有癌症的个体施用有效量的药物。在另一实施方案中,所述药物用于治疗疾病(例如,自身免疫性疾病)、心血管疾病(例如,动脉粥样硬化、高血压、血栓形成)、传染病(例如,埃博拉病毒、马尔堡病毒)或糖尿病的方法中,所述方法包括向个体施用有效量的抗axl抗体或抗体片段。在一个这样的实施方案中,例如,如下所述,该方法还包括向个体施用有效量的至少一种其他治疗剂。在另一实施方案中,所述药物用于抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统(例如肿瘤内脉管系统或肿瘤相关脉管系统)和/或抑制肿瘤基质功能。在另一个实施方案中,所述药物用于在个体中抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统(例如肿瘤内脉管系统或肿瘤相关脉管系统)和/或抑制肿瘤基质功能的方法中,所述方法包括向个体施用有效量的药物以抑制血管生成,抑制细胞增殖,抑制免疫功能,诱导炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统发育(例如肿瘤内脉管系统或肿瘤相关脉管系统发育)和/或抑制肿瘤基质功能。根据任何上述实施方案的“个体”可以是人。
在另一方面,本发明提供了治疗癌症的方法。在一个实施方案中,该方法包括向患有此类癌症的个体施用有效量的抗axl抗体或抗体片段。在一个这样的实施方案中,如下所述,该方法还包括向个体施用有效量的至少一种其他治疗剂。根据任何上述实施方案的“个体”可以是人。
在另一方面,本发明提供治疗免疫疾病(例如,自身免疫性疾病)、心血管疾病(例如,动脉粥样硬化、高血压、血栓形成)、传染病(例如,埃博拉病毒、马尔堡病毒)或糖尿病的方法。在一个这样的实施方案中,如下所述,该方法还包括向个体施用有效量的至少一种其他治疗剂。根据任何上述实施方案的“个体”可以是人。
在另一方面,本发明提供在个体中抑制血管生成,抑制细胞增殖,抑制免疫功能,抑制炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统(例如肿瘤内脉管系统或肿瘤相关脉管系统)和/或抑制肿瘤基质功能的方法。在一个实施方案中,该方法包括向个体施用有效量的抗axl抗体或抗体片段以抑制血管生成,抑制细胞增殖,提高免疫功能,诱导炎性细胞因子分泌(例如,来自肿瘤相关巨噬细胞的炎性细胞因子分泌),抑制肿瘤脉管系统发育(例如肿瘤内脉管系统或肿瘤相关脉管系统发育),和/或抑制肿瘤基质功能。在一个实施方案中,“个体”是人。
在另一方面,本发明提供包含本文提供的任何抗axl抗体或抗体片段的药物制剂,其用于例如,任何上述的治疗方法中。在一个实施方案中,药物制剂包含本文提供的任何抗axl抗体或抗体片段和药学上可接受的载体。在另一个实施方案中,例如,如下所述,药物制剂包含本文提供的任何抗axl抗体或抗体片段和至少一种其他治疗剂。
在上述每个及每种治疗中,本发明的抗体或抗体片段可以单独使用,作为免疫缀合物使用或与治疗中的其他药剂组合使用。例如,本发明的抗体可与至少一种其他治疗剂共同施用。在某些实施方案中,其他治疗剂是抗血管生成剂。在某些实施方案中,其他治疗剂是vegf拮抗剂(在一些实施方案中,是抗vegf抗体,例如贝伐单抗)。在某些实施方案中,其他治疗剂是egfr拮抗剂(在一些实施方案中,是厄洛替尼)。在某些实施方案中,其他治疗剂是化学治疗剂和/或细胞抑制剂。在某些实施方案中,其他治疗剂是紫杉烷(例如紫杉醇)和/或铂剂(例如卡铂)。
在某些实施方案中,其他治疗剂是增强患者免疫力或免疫系统的药剂。
上述组合治疗包括联合施用(其中两种或更多种治疗剂包括在相同或不同的制剂中)和单独施用,在这种情况下,抗体或抗体片段的施用可以在施用其他治疗剂和/或佐剂之前、同时和/或之后进行。抗体或抗体片段也可以与放射治疗组合使用。
可以以符合良好医学实践的方式配制、确定剂量并施用抗体或抗体片段。在此背景下考虑的因素包括所治疗的特定疾病、所治疗的特定哺乳动物、个体患者的临床状况、疾病的原因、药剂的递送部位、施用方法、施用方案以及医生所知的其他因素。抗体或抗体片段不必须地,但任选地与一种或多种目前用于预防或治疗目的疾病的药剂一起配制。这些其他药剂的有效量取决于制剂中存在的抗体或抗体片段的量、疾病的类型或治疗的类型,以及上面讨论的其他因素。这些其他药剂通常以与本文所述的相同的剂量和施用途径使用,或者以本文所述剂量的1%-99%左右使用,或以通过经验/临床确定为适当的任何剂量和任何途径使用。
为了预防或治疗疾病,抗体或抗体片段的适当剂量(当单独使用或与一种或多种其他另外的治疗剂组合使用时)将取决于待治疗的疾病类型、抗体或抗体片段的类型、疾病的严重程度和病程,是否是为了预防或治疗的目的施用抗体或抗体片段、既往治疗史、患者的临床病史和对抗体或抗体片段的反应以及主治医师的判断。抗体或抗体片段适合一次性施用于患者或在一系列治疗中施用于患者。根据疾病的类型和严重程度,无论是例如,通过一次或多次单独的施用,或者通过连续输注,约1μg/kg至40mg/kg的抗体或抗体片段可以是用于向患者施用的初始候选剂量。取决于上述因素,一种典型的日剂量可以为约1μg/kg至100mg/kg或更高。对于数天或更长时间的重复施用,根据情况,通常持续治疗直至出现了所需的对疾病症状的抑制。这些剂量可以间歇施用,例如,每周或每三周施用(例如,使患者接受约2至约20个剂量的抗体或抗体片段,或者例如约6个剂量的抗体或抗体片段)。可以如下进行施用:较高的初始负荷剂量之后以一个或多个较低的剂量施用。然而,其他的给药方案可能是有用的。通过常规技术和测定方法可以较容易地监测治疗的进展。
应当理解,任何上述制剂或治疗方法可以使用本发明的抗体片段或免疫缀合物代替抗axl抗体进行,或者除了抗axl抗体之外还使用本发明的抗体片段或免疫缀合物进行。
增强宿主的免疫功能以对抗肿瘤是越来越受关注的主题。常规方法包括(i)apc增强,例如(a)将编码外源mhc同种异体抗原的dna注入肿瘤,或(b)用增加肿瘤的免疫抗原识别概率的基因转染活检肿瘤细胞(例如,免疫刺激细胞因子,gm-csf,共刺激分子b7.1,b7.2),(iii)过继性细胞免疫疗法,或使用活化的肿瘤特异性t细胞的治疗。过继性细胞免疫疗法包括分离肿瘤浸润性宿主t淋巴细胞,以及例如通过il-2或肿瘤或两者的刺激,在体外扩增群体。另外,分离的功能失调的t细胞也可以通过体外使用抗pd-l1抗体来激活。然后可以将如此激活的t细胞重新施用于宿主。这些方法中的一种或多种可以与施用本发明的抗体、抗体片段或免疫缀合物组合使用。
用于癌症的传统治疗包括以下治疗:(i)放射治疗(例如,放疗,x射线治疗,照射)或使用电离辐射来杀死癌细胞和缩小肿瘤。放射治疗可以通过体外放射治疗(ebrt)在外部进行,或者通过近距离放射治疗在内部进行;(ii)化学治疗,或使用通常影响快速分裂细胞的细胞毒性药物;(iii)靶向治疗,或特异性影响癌细胞中失调蛋白的药剂(例如,酪氨酸激酶抑制剂伊马替尼、吉非替尼;单克隆抗体、光动力治疗);(iv)免疫治疗,或增强宿主的免疫反应(例如疫苗);(v)激素治疗,或激素阻断(例如,当肿瘤对激素敏感时),(vi)血管生成抑制剂,或阻断血管形成和生长,以及(vii)姑息治疗,或旨在改善护理质量的治疗以减轻疼痛、恶心、呕吐、腹泻和出血的治疗。止痛药例如吗啡和羟考酮以及止吐药例如昂丹司琼和阿瑞吡坦可以允许更积极的治疗方案。
在癌症的治疗中,任何先前描述的用于治疗癌症免疫的常规治疗可以在施用抗axl抗体或抗体片段之前、之后或同时进行。此外,抗axl抗体或抗体片段可以在常规癌症治疗之前、之后或同时施用,所述常规癌症治疗包括例如施用肿瘤结合抗体(例如,单克隆抗体,毒素缀合的单克隆抗体)和/或施用化学治疗剂。
f.制品和试剂盒
在本发明的另一个方面,提供了包含可用于治疗、预防和/或诊断上述疾病的物质的制品。制品包括容器以及在容器上或与容器相关的标签或包装说明书。合适的容器包括例如瓶、小瓶、注射器、iv溶液袋等。容器可以由多种材料例如玻璃或塑料制成。所述容器容纳一种组合物,所述组合物自身或者与另一种组合物组合可有效治疗、预防和/或诊断疾病,所述容器可具有无菌入口(例如,所述容器可以是静脉输液袋或具有可被皮下注射针头刺穿的塞子的小瓶)。组合物中的至少一种活性剂是本发明的抗体或抗体片段。标签或包装说明书表明该组合物用于治疗所选择的病症。此外,制品可包括(a)包含组合物的第一容器,其中所述组合物包含抗体或抗体片段;(b)包含组合物的第二容器,其中所述组合物包含另外的细胞毒性剂或其他治疗剂。本发明该实施方案中的制品还可包括包装说明书,其表明该组合物可用于治疗特定病症。作为另外一种选择或除此之外,制品还可包括第二(或第三)容器,该容器包含药学上可接受的缓冲液,例如抑菌性注射用水(bwfi)、磷酸盐缓冲盐水、林格氏溶液和右旋糖溶液。它还可以包括从商业和用户角度考虑所需的其他物品,包括其他缓冲剂、稀释剂、过滤器、针头和注射器。
应理解,任何上述制品可包括代替抗axl抗体的本发明的免疫缀合物或者除了抗axl抗体之外,任何上述制品还可包括本发明的免疫缀合物。
最后,本发明还提供了包含至少一种本发明的抗体或抗体片段的试剂盒。包含本发明的多肽、抗体或抗体片段或抗体药物缀合物的试剂盒可用于检测axl表达(增加或减少),或用于治疗测定或诊断测定。本发明的试剂盒可包含与固体载体偶联的抗体,所述固体载体为例如,组织培养板或珠子(例如琼脂糖珠)。可以提供包含用于在体外(例如,在elisa或western印迹中)对axl进行检测和定量的抗体的试剂盒。可以为这种用于检测的抗体提供标记物,例如萦光标记物或放射性标记物。
试剂盒还包含关于其用途的说明书。在一些实施方案中,说明书包括美国食品和药物管理局对体外诊断试剂盒所要求的说明书。在一些实施方案中,试剂盒还包含用于基于所述样品中存在ax1或不存在ax1来诊断样品中存在脑脊髓液或不存在脑脊髓液的说明书。在一些实施方案中,试剂盒包含一种或多种抗体或抗体片段。在其他实施方案中,试剂盒还包含一种或多种酶、酶抑制剂或酶活化剂。在其他实施方案中,试剂盒还包含一种或多种色谱化合物。在其他实施方案中,试剂盒还包含一种或多种用于制备用于光谱测定的样品的化合物。在进一步的实施方案中,试剂盒还包含对比参考物质,以根据指示剂的强度、色谱或其他物理属性来解释axl的存在或不存在。
以下是本公开的软明胶胶囊的说明性的而非限制性的实施例。对通常在本领域遇到的各种条件和参数进行的对本领域技术人员而言显而易见的其他适当修改和调整都在本发明的范围内。
实施例
实施例1针对axl的条件活性抗体
axl是跨膜酪氨酸激酶,其具有可被条件活性抗体接近的细胞外结构域。该细胞表面蛋白在甲状腺癌组织中高度表达,并在许多其他癌症(例如骨髓增生性疾病、前列腺癌细胞或乳腺癌)中过表达。本文开发了针对axl蛋白的细胞外结构域的条件活性抗体。
选择针对axl的野生型抗体作为模板抗体(具有图1a中063-hum10f10-hc的重链可变区和图1b中的063-hum10f10-hc的轻链可变区)。使用综合位置演化(cpe)演化编码野生型抗体的dna以产生突变抗体文库,通过cpe方法模板抗体中的每个位置一次一个地随机化突变。文库中的每个突变抗体仅具有一个单点突变。如通过elisa测定的,通过在ph6.0与ph7.4下同时筛选对axl的选择性结合亲和力来产生文库中的突变抗体。
同时,突变抗体的表达水平也被优化以用于制造工艺中更高级的领域。使用flag标签在血清中完成筛选,因为血清中存在可能导致筛选的假阳性的人抗体。筛选缓冲液是碳酸盐缓冲液(含有ringer的krebs缓冲液-标准缓冲液,但是不同于pbs)。发现产生的条件活性抗体在ph6.0下对ax1具有更高的亲和力,但在ph7.4下对ax1的亲和力更低,二者都是与野生型抗体相比。一些选择的突变抗体(scfv)如图2所示,它们在ph6.0下比在ph7.4下具有更高的活性,而它们在ph6.0下与ph7.4下的活性比至少为11倍(图3)。
此外,如下表4所示,这些条件活性抗体均具有高表达水平,“克隆”列显示抗体,表达水平“mg/ml”在第二列中显示。
将这些抗体的克隆以及所请求的表达水平(“订购量”,预期表达水平)发送给服务提供者。然而,这些抗体的实际表达水平(“交付量”)非常高并超过预期表达水平。
表4.具有高表达水平的条件活性抗体
以bap063.9-13-1抗体为例,如图4中所示,条件活性抗体在缓冲液中未显示聚集。通过尺寸排阻色谱分析bap063.9-13-1抗体。在图4中,仅检测到一个峰,证明抗体很少聚集或没有聚集。
还使用表面等离子体共振(spr)测定条件活性抗体以测量它们对axl的结合和解离速率。已知spr测定法用于测量条件活性抗体的结合和解离速率。spr测定在碳酸氢盐存在下进行。条件活性抗体的体内结合和解离速率(在动物和人中)是条件活性抗体的非常重要的特征。
与阴性对照(在ph6.0和ph7.4下具有相似结合亲和力的bap06310f10)相比,观察到条件活性抗体在ph6.0下具有更高的结合亲和力而在ph7.4下具有更低的结合亲和力(图5)。此外,将温度从室温升至60℃不会明显改变elisa测定结果(图5)。elisa测定还显示,与在ph7.4下相比,这些条件活性抗体在ph6.0下具有高选择性(图6a-6b显示一种抗体作为实例)。
条件活性生物抗体总结于表5中。这些抗体中的两种表达为scfv(bap063.9-13.3和bap063.9-48.3)。将抗体在60℃孵育1小时不会改变大多数抗体的亲和力(“热稳定性”)。
条件活性抗体可用于根据本发明检测ctc表面上的axl蛋白。
表5-条件活性抗axl抗体的总结
实施例2:抗axl抗体的ph依赖性结合亲和力
在不同ph水平的缓冲液中测试了本发明的一些抗axl抗体。一种缓冲液是含有1%牛血清白蛋白(bsa)的krebs缓冲液。滴定krebs缓冲液使ph值在5-7.4范围内。使用elisa测定法(od450)测量抗体与axl的结合亲和力,结果显示在图7中。两种对照抗体(bap063-3831和bap063-3818)没有条件活性,因为它们的结合亲和力受ph变化影响不明显。另一方面,本发明的抗axl抗体具有条件活性,因为它们与axl的结合亲和力取决于ph(图7)。
实施例3:用抗axl抗体进行的细胞杀伤
使用a549细胞测试本发明的抗axl抗体的细胞杀伤活性。结果显示在图8a-8e中。在两个ph水平:6.0和7.4(分别代表肿瘤微环境中的ph和正常的生理ph)下,测量细胞杀伤活性。各种抗体浓度下的细胞杀伤百分比如图8a-8e所示。
这两种测试给出了一致的细胞杀伤结果。阴性对照(抗axl人源化wt)在ph6.0下和ph7.4下对a549细胞显示出相似的细胞杀伤活性(图8a)。相反,与ph7.4下的细胞杀伤活性相比,本发明的抗axl抗体在ph6.0下显示出明显更高的细胞杀伤活性,特别是在抗体相对于a549细胞并不饱和的低抗体浓度下(图8b-8e)。
实施例4:抗axl抗体对cyno-axl的结合亲和力
在ph6.0和ph7.4下的两种不同缓冲液中,测量本发明的抗axl抗体对cyno-axl的结合亲和力,并与对人axl(hax1)的结合亲和力进行比较。结果显示在图9a-9d中。cyno-axl是来自非人灵长类动物即食蟹猴(cynomolgusmacaquesmonkey)的axl蛋白。
在ph6.0和ph7.4下的两种缓冲液中,对照品(ba-3831-wt)均显示出对人axl(hax1)和对食蟹猴axl(cyno-axl)的相似的结合亲和力(图9a)。本发明的抗axl抗体在两种缓冲液之一中显示出对hax1和猕猴-axl相似的结合亲和力谱,即与在ph6.0下对cyno-axl的结合亲和力相比,在ph7.4下对cyno-axl的结合亲和力较低(图9b-9d)。在另一种缓冲液中,ph6.0下和ph7.0下结合亲和力之间的差异不明显。
实施例5:与金霉素缀合的抗axl抗体的细胞毒性
金霉素具有细胞毒性,因为它通过阻止蛋白质合成来抑制细胞生长。将本发明的抗axl抗体之一,bap063.94007,与金霉素缀合。在该测试中使用两种对照品,bap063humwt和b12(抗b12抗体),两者也是与金霉素缀合。
在ph6.0下和ph7.4下,用三种金霉素缀合的抗体(bap063.94007,bap063humwt和b12)处理几种细胞系(图10a-10h)。本发明的金霉素缀合的抗体bap063.94007在ph6.0下对细胞系du145(前列腺癌细胞)、mda-md-231(乳腺癌细胞)、pl45(胰腺癌细胞)和a549(腺癌细胞)显示出与ph7.4下对相同细胞的细胞毒性相比明显更高的细胞毒性。
实施例6:与模型毒素缀合的抗axl抗体
将本发明的抗axl抗体与模型毒素(例如吉西他滨)缀合以产生条件活性抗体-药物缀合物(cab-axl-adc)。首先测试cab-axl-adc以确认药物缀合过程未改变条件细胞杀伤活性。该测试显示,与ph7.4下相比,cab-axl-adc在ph6.0下杀死了明显更多的细胞(图11)。
然后将cab-axl-adc以1mg/kg的剂量(每周两次,持续3周)注射到携带miapaca2异种移植肿瘤的小鼠中。在该研究中使用了几种对照品,包括裸cab(裸条件活性生物制剂,即没有进行缀合的抗axl抗体)、载体、单独的毒素(未缀合的吉西他滨)、对照adc、亲和力匹配(affinitymatching)的抗axladc(amadc)。该研究表明,与对照品相比,cab-axl-adc(cabadc)和amadc明显使肿瘤尺寸减少更多(图12)。未缀合的抗axl抗体不会减小肿瘤的尺寸。该研究表明,与毒素缀合的抗axl抗体与亲和力匹配抗体一样可以有效地减小肿瘤的尺寸。
实施例7:食蟹猴中抗axl抗体药物缀合物的血清浓度
将本发明的抗axl抗体的药物(金霉素)缀合物(cab-adc)以三种剂量注射到雄性猕猴和雌性食蟹猴中:0.1mg/kg、1mg/kg和10mg/kg。用裸抗axl抗体作为对照品。亲和力匹配抗体药物缀合物(am-adc)也用作对照品。在一周的时间内测量抗体的血清浓度(168小时,参见图13a-13b)。cab-adc在猴血清中持续的时间比am-adc对照品更长(图13b)。雄性猴和雌性猴之间没有明显差异(图13a-13b)。
实施例8:抗axl抗体药物缀合物在食蟹猴中的毒性
测试了本发明的cab-adc在食蟹猴中的毒性。天冬氨酸转氨酶(ast)和丙氨酸转氨酶(alt)已被美国食品和药物管理局(fda)用作药物的肝毒性指标。测量了雄性猴和雌性猴的血清ast水平和alt水平(图14a-14b)。载体(pbs)在血清中未引起ast或alt水平改变,而匹配抗体药物缀合物(am)在施用10mg/kg剂量后3天显示出非常高的ast水平和alt水平。与am对照品相比,cab-adc显示出显著降低的ast水平和alt水平。这表明相对于匹配抗体药物缀合物am,本发明的抗axl抗体药物缀合物具有显著降低的肝毒性。
还发现本发明的抗axl抗体药物缀合物在猴子中引起较少的炎症(图15)。在注射cab、am和pbs后,收集猴子血液中的淋巴细胞计数。与引起明显炎症的am相比,本发明的抗axl抗体药物缀合物(cab-adc)仅在猴子中引起轻度炎症。
实施例9:小鼠体内实验
将可发展成肿瘤的2种肿瘤细胞系(lclc103h或du145)中的一种植入小鼠。测量用抗肿瘤药物一甲基奥利斯他汀e(mmae)处理后的肿瘤尺寸。对于接受lclc103h的小鼠,用单剂量的载体(作为阴性对照)、cab抗axl抗体缀合的mmaeadc(cabaxl-mmae)或非-cab抗axl抗体缀合的mmaeadcc(non-cabaxl-mmae)处理小鼠(图16a)。用adc治疗的小鼠中的肿瘤缩小,而用载体治疗的小鼠中的肿瘤继续生长。
此外,用载体(阴性对照)或cab抗axl抗体缀合的mmaeadc(cabaxl-mmae)以两种不同浓度(6mg/kg和10mg/kg)处理接受du145的小鼠。随时间测量肿瘤体积。肿瘤在阴性对照组(载体)中继续生长,而在用adc处理的小鼠中肿瘤生长减慢(图16b)。
然而,应该理解,尽管在前面的描述中已经阐述了本发明的许多特征和优点以及本发明的结构和功能的细节,但是本公开仅是说明性的,并且可以在表达所附权利要求的术语的广泛一般含义所指示的全部范围内具体地进行改变,特别是在本发明原理内在形状、尺寸和部件布置方面进行改变。
本文提及的所有文献均通过引用整体并入本文,或者替代地提供它们所具体依据的公开内容。申请人无意将任何公开的实施方案提供给公众,因此从这种意义上说,任何公开的修改或变更可能字面上不落入权利要求的范围内,但它们在等同原则下被认为是本文的一部分。
- 还没有人留言评论。精彩留言会获得点赞!