体外肾毒性筛选测定法的制作方法
本发明涉及使用测量作为毒性生物标志物的表皮生长因子(egf)水平(潜在地结合其他生物标志物,如三磷酸腺苷(atp)和肾损伤分子-1(kim-1))的体外基于细胞的测定法预测药物(特别是核酸分子,如sirna或反义寡核苷酸)的体内肾毒性的方法。本发明进一步涉及使用所述测定法从药物(特别是核酸分子,人rnai剂或反义寡核苷酸)文库选择用于体内给药的一种或多种药物的方法。发明背景在鉴定新的以及优化的药物候选物中的问题之一是剂量限制性毒性事件。在体内评价时,通常有药物化合物子集将引发毒性表型,如药物诱发的肾损伤或肾毒性。在过去几年中,已经使用各种生物标志物研发了用于预测药物肾毒性的许多模型。sohn等2013toxicologylettersvol217pp235和huang等2015pharmacologicalresearch&perspectivesvol3ppe00148都公开了使用包括肾损伤分子-1(kim-1)的各种生物标志物预测小分子的肾毒性的体外基于细胞的测定法。ju等2015sciencetranslationalmedicinevol7,pp316ra193描述了表皮生长因子(egf)作为针对慢性肾病的潜在体内生物标志物,通过测量与肾小球滤过率相关的尿液中的egf转录物和分泌。没有描述体外测量胞外egf摄入。wilmer等2016trendsinbiotechnologyvol34pp156综述了用于体内和体外筛选药物诱发的肾毒性的更多生物标志物(参见表2)。体外肾毒性测定法已经显示出一定的预测一些小分子或多肽药物的已知肾毒性的能力,所述小分子或多肽药物如两性霉素b(抗真菌剂)、粘菌素(多肽抗生素)、环孢菌素(免疫抑制剂)、顺铂(化疗剂)、阿霉素(化疗剂)、庆大霉素(抗细菌剂)。然而,还不存在针对不同类别的药物(即基于核酸的药物,如irna、反义寡核苷酸和适体)的情况。之前已经公开了单个核酸化合物的肾毒性并且显示出是不可预测的(参见例如henry等1997toxicologyvol120pp145;monteith等1999toxicologicpathologyvol27,pp.307;herrington等2011americanjournalofkidneydiseases:theofficialjournalofthenationalkidneyfoundationvol57,pp300;voit等2014thelancet.neurologyvol13,pp987;vanpoelgeest等2015britishjournalofclinicalpharmacologyvol80,pp1350)。据我们所知,使用egf作为生物标志物用于预测药物的肾毒性的体外基于细胞的测定法尚未有描述。特别地,基于核酸的分子的肾毒性的体外预测之前也没有描述。发明目的本发明确定了egf作为体外基于细胞的测定法中可靠的生物标志物,所述测定法用于预测药物化合物的体内肾毒性,特别是用于核酸分子,如反义寡核苷酸。可靠的肾毒性的体外预测将提高成功的药物临床研发,并且在针对最初的筛选药物发现中无需使用动物将更加节约成本、有效且合乎伦理,显著减少药物文库的毒性筛选所需的动物数量。发明概述本发明提供了体外毒性测定法,已经发现了其可用于预测药物的体内肾毒性,所述药物特别是寡核苷酸,如反义寡核苷酸。在本发明的一个方面中,本发明的发明人已经鉴定了将药物给药于表达表皮生长因子受体(egfr)的细胞时,表皮生长因子(egf)作为肾毒性的生物标志物。egf生物标志物可以结合其他生物标志物,如三磷酸腺苷(atp)和/或肾损伤分子-1(kim-1)。本发明提供了用于预测体内毒性(或体内毒性可能性)的体外方法(测定法),其可以用于选择适用于或预测适用于体内给药而没有不利的肾毒性(如急性肾损伤或药物诱发的肾损伤)的药物化合物。本发明提供了用于预测药物(特别是基于核酸的分子,如哺乳动物中的反义寡核苷酸)的体内肾毒性的方法,所述方法包括步骤:a)在含有至少4ng/ml表皮生长因子(egf)的合适细胞培养基中培养表达表皮生长因子受体(egfr)的细胞;b)将药物给予所述细胞培养物;c)孵育细胞一段时间;和d)随后测量上清液中的egf水平;其中上清液中的egf的提高表示药物是或预测是与肾毒性相关。本发明提供了从药物文库选择一种或多种适用于体内给药的药物的方法,所述方法包括步骤:a.获得药物文库;b.将药物文库的每个成员给予表达表皮生长因子受体(egfr)的细胞培养物并且其中培养基含有至少4ng/ml的表皮生长因子(egf);c.体外培养细胞一段时间;d.针对每种药物测量胞外egf的含量;和e.选择一种或多种其中毒性等级低于6的药物。任选地,所述方法可以进一步包括将选定的药物体内施与哺乳动物的步骤。合适地,在本发明的方法中,可以将至少一种生物标志物的水平或含量与给药无毒参照药物(证实在体内是无毒的)时获得的水平进行比较,以确定至少一种生物标志物的提高或降低的水平是由于药物的给药引起的(即,肾毒性的至少一种生物标志物的改变)。此外,可以将至少一种生物标志物与给药毒性参照药物(证实在体内是有毒的)时获得的水平进行比较,以确定至少一种生物标志物的测定窗口。在一些实施方案中,表达egfr的细胞选自源自上皮细胞、间充质细胞、神经外胚层细胞和肝细胞的细胞培养物。特别地,源自肾上皮细胞的细胞培养物在本发明的方法中是有用的,如人ptec或人ptec-tert-1细胞培养物。在一些实施方案中,预测的肾毒性与细胞培养基中作为生物标志物的egf的提高有关。在进一步的实施方案中,kim-1水平的提高表示药物是或预测是与肾毒性相关。用于预测肾毒性的更多生物标志物可以与胞内atp水平的降低有关。本发明提供了体外测定法用于确定药物的肾毒性(例如,可能性)的用途,所述药物特别是基于核酸的分子,如寡核苷酸,如lna寡核苷酸。本发明提供了通过本发明的用于预测肾毒性的方法或通过用于筛选药物分子文库的方法获得的药物。本发明提供了包含通过本发明的用于预测肾毒性的方法或通过用于筛选药物文库的方法获得的药物和可药用稀释剂、溶剂、运载体、盐和/或佐剂的药物组合物。本发明提供了通过本发明的用于预测肾毒性的方法或通过用于筛选药物文库的方法获得的核酸分子或包含这样的分子的药物组合物,用于药物中。附图简述图1显示了用100μm寡核苷酸处理7天的ptec-tert1细胞的形态学变化。图2图示了各种生物标志物怎样与体内观察到的毒性关联。白色表示无毒性(无害的);浅灰色表示轻度毒性;中灰色表示中等毒性;和深灰色表示高毒性。定义药物本申请内容中的术语“药物”通常理解为用于疾病或病症的治疗、减轻或预防的活性成分。药物还可以理解为包含活性成分的组合物,例如,药物组合物。通常,可对任何药物实施本发明的体外肾毒性预测方法。在一些实施方案中,药物选自基于核酸的分子、抗癌剂;氨基糖苷类;抗细菌剂、抗病毒剂;抗真菌剂、抗炎剂和免疫抑制剂。具有已知肾毒性的抗细菌剂的实例是多粘菌素,如粘菌素。在一些实施方案中,药物是选自rnai剂、反义寡核苷酸和适体的核酸分子。在一些实施方案中,药物是反义寡核苷酸。药物文库理解为具有共同的可识别的化学结构的药物的集合。通常,药物文库是基于期望对其进行改进的亲本或祖先药物,例如,在本申请的内容中,已经发现亲本药物引发体内肾毒性。无限增殖化细胞系本技术实现要素:中的术语“无限增殖化细胞系”理解为从多细胞生物体的单个细胞遗传得到的细胞群,其中细胞已经得到修饰,以逃避正常的细胞衰老,允许细胞连续增殖或分裂。无限增殖化细胞可以在体外生长延长的时间段。用于永生所需的突变可以是天然产生的或有意诱导的,例如,使用病毒载体、基因删除、基因诱导或蛋白质(如端粒酶)诱导或与永生细胞(如癌细胞)融合。原代细胞培养物本
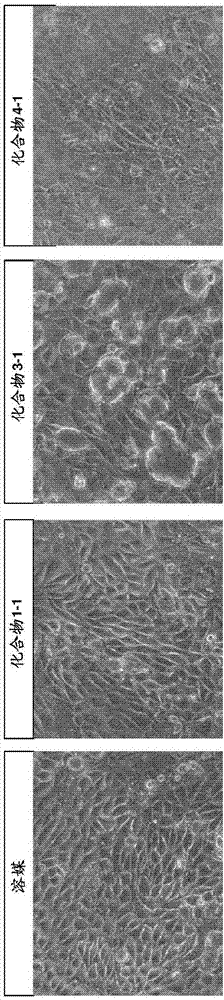
发明内容中使用的术语“原代细胞培养物”理解为从动物中的特定组织分离的细胞群。没有之前的遗传操纵或克隆选择来培养原代细胞培养物,然而可以使用例如facs或选择生长条件来纯化。原代细胞培养物可以是新鲜的或从低温保存的组织建立。靶核酸根据本发明,靶核酸可以是编码哺乳动物蛋白的核酸,并且可以例如是基因、rna、mrna和前-mrna、成熟mrna或cdna序列。靶核酸还可以是微rna、长的非编码rna、小的核仁rna或转移rna。核酸分子如本文中使用的术语“核酸分子”或“治疗性核酸分子”按照本领域技术人员通常理解的定义为包含两个或更多个共价连接的核苷的分子。本发明的方法中涉及的核酸分子通常是长度低于50个核苷酸的治疗性寡核苷酸。核酸分子可以是或包含反义寡核苷酸,或可以是另一个寡聚核酸分子,如rnai剂、适体或核酶。核酸分子通常是在实验室中通过固相化学合成接着纯化制得的。提及核酸分子的序列时,是指共价连接的核苷酸或核苷的核碱基部分的序列或顺序,或其修饰。本发明的核酸分子是人造的,并且是化学合成的,并且通常是纯化或分离的。本发明的核酸分子可以包含一个或多个修饰的核苷或核苷酸。在一些实施方案中,本发明的核酸分子包含8至40个核苷酸长度或由8至40个核苷酸长度组成,如9至35,如10至30,如11至22,如12至20,如13至18,或14至16个连续的核苷酸长度。在一些实施方案中,核酸分子或其连续的核苷酸序列包含22个或更少的核苷酸,或由22个或更少的核苷酸组成,如20个或更少的核苷酸,如18个或更少的核苷酸,如14、15、16或17个核苷酸。将理解本文中给出的任何范围包括范围端点。因此,如果说核酸分子包括10至30个核苷酸,则包括10个和30个核苷酸。在一些实施方案中,连续的核苷酸序列包括8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个连续的核苷酸长度,或由8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个连续的核苷酸长度组成。核酸分子通常用于调节哺乳动物中的一个或多个靶核酸的表达。在一些实施方案中,核酸分子,如sirna和反义寡核苷酸,通常用于抑制哺乳动物中的rna(例如,如mrna或微rna)的表达。核酸分子因此在调节哺乳动物中的一个或多个靶核酸的表达中是有效的。在本发明的一个实施方案中,核酸分子选自rnai剂、反义寡核苷酸或适体。在一些实施方案中,核酸分子是硫代磷酸酯核酸分子。在一些实施方案中,核酸分子包含硫代磷酸酯核苷间键。在一些实施方案中,核酸分子可以与非核苷部分(缀合物部分)缀合。在一些实施方案中,本发明的方法中使用或鉴定的核酸分子包含至少一个立体限定的硫代磷酸酯核苷间键。核酸分子文库理解为变体核酸分子的集合。核酸分子文库的目的可以不同。在一些实施方案中,核酸分子文库由具有不同核碱基序列的寡核苷酸组成,例如,其可以是跨越靶核酸(例如,rna序列)设计的核酸分子的文库,例如,通过mrna基因行走(gene-walk)产生的反义寡核苷酸或rnai剂的文库,目的在于鉴定靶核酸上核酸分子有效调节靶核酸的区域。在一些实施方案中,核酸分子文库由具有靶向靶核酸上特定区域的重叠核碱基序列的寡核苷酸组成,目的在于鉴定核酸分子文库内最有效的序列。在一些实施方案中,核酸分子的文库是亲本或祖先核酸分子的核酸分子设计变体(子核酸分子)的文库,其中核酸分子设计变体保留亲本核酸分子的核心核碱基序列。在一些实施方案中,核酸分子变体(子核酸分子)的文库与亲本核酸分子的一个或多个设计参数不同。此类文库的目的在于改进亲本核酸分子,例如,在本申请的内容中,已经发现亲本核酸分子在体内引发肾毒性。反义寡核苷酸如本文中使用的“反义寡核苷酸”定义为能够通过与靶核酸(特别是与靶核酸上的连续序列)杂交来调节靶基因表达的核酸分子。反义寡核苷酸通常含有一个或多个dna或dna样核苷段。反义寡核苷酸不一定是双链的并且因此不是sirna。优选地,本发明的反义寡核苷酸是单链的。在一些实施方案中,反义寡核苷酸能够募集rnaseh,并且可以例如是如本文中定义的缺口聚物寡核苷酸,在侧翼包含一个或多个2’糖修饰的核苷,如2’-o-烷基-rna、2’-o-甲基-rna、2’-烷氧基-rna、2’-o-甲氧基乙基-rna、2’-氨基-dna、2’-氟-dna、阿糖(arabino)核酸(ana)、2’-氟-ana和lna核苷或这些的混合物(混合的翼缺口聚物),或者可以是缺口打破者(gap-breaker)寡核苷酸。在一些实施方案中,反义寡核苷酸是混合聚物(mixmer)。混合聚物寡核苷酸通常包含高亲和性2’糖修饰的核苷(如2’-o-烷基-rna、2’-o-甲基-rna、2’-烷氧基-rna、2’-o-甲氧基乙基-rna、2’-氨基-dna、2’-氟-dna、阿糖核酸(ana)、2’-氟-ana和lna核苷)与1-4或1-3个dna核苷的短区域的交替区域。通常混合聚物包含具有短dna段的交替区域,例如,[lna]1-5[dna]1-3[lna]1-4[dna]1-3[lna]1-4[dna]1-3。多种混合聚物设计是高度有效的,例如,当靶向微rna(反义mirs)、mrna上的微rna结合位点(blockmirs)或作为剪接转换寡聚物(aso)时。参见例如wo2007/112754(lna-antimirstm),wo2008/131807(lna剪接转换寡聚物)。在一些实施方案中,寡核苷酸可以是7-10个核苷酸长度的tinylna寡核苷酸。这样的tinylna公开于wo2009/043353中,在此并入本文作为参考。它们通常用于抑制微rna和微rna家族,并且可以是全lna修饰的(即,每个核苷都是lna核苷)。还优选与缺口聚物和混合聚物寡核苷酸一样,核苷间键包含硫代磷酸酯核苷间键,并且与本文中提及的寡核苷酸一样,可以是完全的硫代磷酸酯寡核苷酸。反义寡核苷酸通常为7-30个核苷酸长,如7-10个核苷酸(例如,tinylna)或10-14个核苷酸(例如,短聚物(shortmer)或短缺口聚物)或12-20或10-22或10-24个核苷酸长。irna如本文中使用的,术语“rnai”、“rnai剂”、“irna剂”、“rna干扰剂”或“sirna”在本文中可互换使用,是指含有rna核苷并且介导靶核酸(如rna转录产物)经由rna诱导的沉默复合物(risc)途径靶向切割的寡核苷酸分子。irna指引mrna通过称为rna干扰(rnai)的过程的序列特异性降解。irna调节(例如,抑制)细胞(例如,受试者(如哺乳动物受试者)体内的细胞)中的靶核酸的表达。实施本发明方法的rnai剂可以是与靶核酸序列(如靶rna序列)相互作用的双链rna(dsrna)或单链rna分子,通过risc途径来指引靶核酸的切割。双链rnai剂通常为20至50个核苷酸长,如25至35个核苷酸长,并且与称为dicer的核酸内切酶相互作用,认为dicer将dsrna加工成19-23个碱基对的短干扰rna,具有特征性的两个3’突出碱基,其随后结合至rna-诱导的沉默复合物(risc)中。双链rnai剂可以是由一起形成双链体的正义链和反义链组成的sirna分子。备选地,可以是形成二级发夹结构的寡核苷酸,所述二级发夹结构实质性地使其在发夹区域是双链的,这样的分子也称为shrna。有效的延伸形式的dicer底物已经描述于us8,349,809和us8,513,207中,在此并入本文作为参考。结合合适的靶mrna时,risc内的一个或多个核酸内切酶将靶切割,以诱导沉默。单链rnai剂结合risc核酸内切酶argonaute2,其随后切割靶mrna。单链irna通常为15-30个核苷酸长并且是化学修饰的,例如,包括修饰的核苷间键,并且还可能是修饰的核苷。单链sirna的设计和测试描述于美国专利号8,101,348和lima等(2012)celli50:883-894中,在此并入本文作为参考。dsrna可以用单链rnai剂相同的方式进行化学修饰。适体如本文中使用的,术语适体是指形成能够通过配体-靶相互作用(如配体-蛋白相互作用或配体-dna螺旋体相互作用)调节靶的三维结构的寡核苷酸或肽。寡核苷酸适体可以由dna、rna或修饰的核苷或这些的混合物组成。适体通过其三维结构是有效的,而不是通过反义寡核苷酸或rnai剂那样的靶杂交。修饰的核苷间键修饰的核苷间键可以例如选自硫代磷酸酯、二硫代磷酸酯(diphosphorothioate)和硼烷磷酸酯(boranophosphate)。在一些实施方案中,修饰的核苷间键与本发明方法中待测试的寡核苷酸的rnaseh募集相容,例如,硫代磷酸酯、二硫代磷酸酯或硼烷磷酸酯。在一些实施方案中,核苷间键包含硫(s),如硫代磷酸酯核苷间键。在一些实施方案中,本发明的方法中使用或鉴定的寡核苷酸包含至少一个立体限定的硫代磷酸酯核苷间键。由于核酸酶抗性、有益的药物动力学和易于制造,硫代磷酸酯核苷间键特别有用。在一些实施方案中,寡核苷酸或其连续的核苷酸序列中至少50%的核苷间键是硫代磷酸酯,如寡核苷酸或其连续的核苷酸序列中至少60%,如至少70%,如至少80或如至少90%的核苷间键是硫代磷酸酯。在一些实施方案中,寡核苷酸或其连续的核苷酸序列的全部核苷间键是硫代磷酸酯。在一些实施方案中,寡核苷酸包含一个或多个中性核苷间键,特别是选自磷酸三酯、膦酸甲酯、mmi、酰胺-3、甲缩醛(formacetal)或硫代甲缩醛的核苷间键。更多的核苷间键公开于wo2009/124238中(在此并入本文作为参考)。在一个实施方案中,核苷间键选自wo2007/031091中公开的接头(在此并入本文作为参考)。特别地,核苷间键可以选自-o-p(o)2-o-、-o-p(o,s)-o-、-o-p(s)2-o-、-s-p(o)2-o-、-s-p(o,s)-o-、-s-p(s)2-o-、-o-p(o)2-s-、-o-p(o,s)-s-、-s-p(o)2-s-、-o-po(rh)-o-、o-po(och3)-o-、-o-po(nrh)-o-、-o-po(och2ch2s-r)-o-、-o-po(bh3)-o-、-o-po(nhrh)-o-、-o-p(o)2-nrh-、-nrh-p(o)2-o-、-nrh-co-o-、-nrh-co-nrh-,和/或核苷间接头可以选自:-o-co-o-、-o-co-nrh-、-nrh-co-ch2-、-o-ch2-co-nrh-、-o-ch2-ch2-nrh-、-co-nrh-ch2-、-ch2-nrhco-、-o-ch2-ch2-s-、-s-ch2-ch2-o-、-s-ch2-ch2-s-、-ch2-so2-ch2-、-ch2-co-nrh-、-o-ch2-ch2-nrh-co-、-ch2-nch3-o-ch2-,其中rh选自氢和c1-4-烷基。与靶核酸(如用于缺口聚物(gapmer)的区域g,或头聚物(headmer)和尾聚物(tailmer)的非修饰核苷区)形成双链体时,核酸酶抗性键,如硫代磷酸酯键,在能够募集核酸酶的寡核苷酸区域中特别有用。然而,硫代磷酸酯键在非核酸酶募集区和/或亲和性增强区中也是有用的,如用于缺口聚物的f和f’区,或头聚物和尾聚物的修饰核苷区。然而,每个设计区包含除了硫代磷酸酯键以外的其他核苷间键,如磷酸二酯键,特别是在其中修饰的核苷(如lna)保护对抗核酸酶降解的键的区域中。包含磷酸二酯键,如一个或两个键,特别是在修饰的核苷单体之间或邻近修饰的核苷单体(特别是在非核酸酶募集区中),可以改变寡核苷酸的生物利用率和/或生物分布—参见wo2008/113832,在此并入本文作为参考。在一个实施方案中,寡核苷酸中的所有核苷间键是硫代磷酸酯和/或硼烷磷酸酯键。在一些实施方案中,寡核苷酸中的所有核苷间键是硫代磷酸酯键。立体限定的核苷间键在本发明的内容中,术语“立体限定的”是指其中寡核苷酸中存在的至少一个硫代磷酸酯核苷间键具有限定的立体化学(即,rp或sp)的核酸分子,如包括反义、rni和适体分子的寡核苷酸。在一些实施方案中,立体限定的寡核苷酸中的所有硫代磷酸酯核苷间键可以是立体限定的,即,每个硫代磷酸酯核苷间键独立地选自rp和sp硫代磷酸酯核苷间键。通常,作为rp和sp硫代磷酸酯键的随机混合物(也称为外消旋混合物)来合成寡核苷酸硫代磷酸酯。在本发明中,提供了缺口聚物硫代磷酸酯寡核苷酸,其中寡核苷酸样品中存在的至少75%,如至少80%,或至少85%,或至少90%或至少95%,或至少97%,如至少98%,如至少99%,或(实质上)全部的寡核苷酸分子中的缺口区寡核苷酸的至少一个硫代磷酸酯键是立体限定,即rp或sp。这样的寡核苷酸可以称为是立体限定的、立体选择性的或立体特异性的(stereospecified):它们包含至少一个是立体特异性的硫代磷酸酯键。术语立体限定的和立体特异性的/立体选择性的在本文中可互换使用。术语立体限定的、立体选择性的和立体特异性的可以用于描述硫代磷酸酯键(rp或sp),或可以用于描述包含这样的硫代磷酸酯核苷间键的寡核苷酸。认识到立体限定的寡核苷酸可以在任一个位置包含少量的可替换的立体异构体,例如,wan等在nar,2014年11月中报道了98%的立体选择性,用于报道的缺口聚物。表达的调节如本文中使用的术语“表达的调节”理解为与给予寡核苷酸前的核酸靶含量相比时寡核苷酸改变核酸靶含量的能力的统称。备选地,可以通过参照对照实验来测定表达的调节。通常理解对照是用盐水组合物处理的个体或靶细胞或用非靶向寡核苷酸(模拟物)处理的个体或靶细胞。然而,还可以是用标准护理处理的个体。一种类型的调节是寡核苷酸抑制、下调、降低、遏制、去除、停止、阻断、防止、减少、缩减、避免或终止核酸靶表达的能力,例如,通过mrna的降解或转录的阻断。另一种类型的调节是寡核苷酸恢复、提高或增强核酸靶表达的能力,例如,通过修复剪接位点或防止剪接或去除或阻断抑制机制,如微rna抑制。在本发明的一些实施方案中,本发明的寡核苷酸的靶存在于egfr表达细胞中时,本发明的方法可以进一步包括用寡核苷酸处理后(例如,这可以与至少一个生物标志物测量步骤平行进行或作为其一部分进行)测定egfr表达细胞群中的靶调节水平(例如,抑制sirna或反义寡核苷酸)的步骤。在这点上,本发明的方法可以用于测定寡核苷酸的比较功效或有效性和比较毒性,允许选择有效的无毒化合物用于体内。将理解可以在分开的体外实验中,在egfr表达细胞中,特别是正在表达靶的细胞中,进行化合物功效/有效性的测定。修饰的寡核苷酸未修饰的dna和rna在体内快速降解,并且其本身在治疗上很少有用途。通常,因此本发明的方法中使用的寡核苷酸是修饰的。一种广泛使用的修饰是使用硫代磷酸酯核苷间键,已知其使寡核苷酸稳定,以免核酸水解降解,以及提供理想的药理学特性。在一些实施方案中,寡核苷酸包含硫代磷酸酯核苷间键。另一种理想的修饰是给予寡核苷酸对靶核酸较高亲和性的那些,因此称为高亲和性修饰的核苷酸,其包括双环“lna”核苷以及许多2’取代的核苷。高亲和性修饰的核苷在一些实施方案中,寡核苷酸包含一个或多个高亲和性修饰的核苷。高亲和性修饰的核苷是结合至寡核苷酸中时增强寡核苷酸对其互补靶的亲和性的修饰核苷,例如通过熔化温度(tm)测量的。本发明的高亲和性修饰的核苷优选导致每个修饰的核苷的熔化温度提高+0.5至+12℃,更优选+1.5至+10℃,并且最优选为+3至+8℃。各种高亲和性修饰的核苷是本领域已知的并且包括例如2’糖修饰的核苷,如许多2’取代的核苷,以及锁核酸(lna)(参见,例如,freier&altmann;nucl.acidres.,1997,25,4429-4443和uhlmann;curr.opinionindrugdevelopment,2000,3(2),293-213)。糖修饰寡核苷酸可以包含一个或多个具有修饰的糖部分的核苷,即,与天然产生的dna和rna核苷中发现的核糖糖部分相比时糖部分的修饰。已经制得了具有核糖糖部分修饰的各种核苷,主要目的在于提高寡核苷酸的某些特性,如亲和性和/或核酸酶抗性。这样的修饰包括其中核糖环结构被修饰的那些,例如,用己糖环替代(hna)。糖修饰还包括通过将核糖环上的取代基变成除了氢或dna和rna核苷中天然产生的2’-oh基团以外的其他基团进行的修饰。例如,可以在核糖环的2’、3’、4’或5’位置引入取代基。具有修饰的糖部分的核苷还包括2’修饰的核苷,如2’取代的核苷。已经发现各种2’取代的核苷在引入寡核苷酸中时具有有益的特性,如增强的核苷抗性和增强的亲和性。在一个实施方案中,核酸分子或此类分子的文库包含一个或多个2’糖修饰的核苷。除了2’取代,存在其他修饰,包括核糖环的位置2的修饰,如引入双环,其通常在核糖环上的c2和c4碳之间具有双游离基(biradicle)桥(也称为锁核酸或lna),或未连接的核糖环,其通常在c2和c3碳之间缺少键(例如,una)。其他糖修饰的核苷包括例如双环己糖核酸(wo2011/017521)或三环核酸(wo2013/154798)。修饰的核苷还包括其中糖部分被非糖部分替代的核苷,例如,在肽核酸(pna)或吗啉代核酸的情况中。2’糖修饰的核苷2’糖修饰的核苷是在2’位置具有除了h或-oh以外的取代基(2’取代的核苷)或包含2’连接的双游离基的核苷,并且包括2’取代的核苷和lna(2’-4’双游离基桥)核苷。例如,2’修饰的糖可以给核苷酸提供增强的结合亲和性和/或提高的核酸酶抗性。2’取代的修饰的核苷的实例是2’-o-烷基-rna、2’-o-甲基-rna、2’-烷氧基-rna、2’-o-甲氧基乙基-rna(moe)、2’-氨基-dna、2’-氟-rna和2’-f-ana核苷。对于更多的实例,请参见例如freier&altmann;nucl.acidres.,1997,25,4429-4443和uhlmann;curr.opinionindrugdevelopment,2000,3(2),293-213,以及deleavey和damha,chemistryandbiology2012,19,937。以下是一些2’取代的修饰的核苷的说明。锁核酸核苷(lna)在一些实施方案中,寡核苷酸是lna寡核苷酸,即,它们包含至少一个lna核苷。lna单体(也称为双环核酸,bna)是其中在核糖环的2’和4’位置之间存在双游离基的核苷。2’-4’双游离基也称为桥。lna单体,当掺入至寡核苷酸中时,已知能增强寡核苷酸与互补dna或rna序列的结合亲和性,通常按照熔化寡核苷酸/靶双链体需要的温度(tm)的提高来测量或计算。lna寡聚物可以是单链反义寡核苷酸。本发明的寡核苷酸化合物中使用的lna具有通式i的结构其中对于所有手性中心,可以发现r或s方向的不对称基团;其中x选自-o-、-s-、-n(rn*)-、-c(r6r6*)-,如,在一些实施方案中,-o-;b选自氢、任选取代的c1-4烷氧基、任选取代的c1-4烷基、任选取代的c1-4酰氧基、包括天然产生的和核碱基类似物的核碱基、dna嵌入剂、光化学活性基团、热化学活性基团、螯合基团、报告基团和配体;优选地,b是核碱基或核碱基类似物;p表示邻近单体的核苷酸间键,或5’端基,这样的核苷酸间键或5’端基任选包括取代基r5或同样适用的取代基r5*;p*表示邻近单体的核苷酸间键,或3’端基;r4*和r2*一起表示由1-4个选自-c(rarb)-、-c(ra)=c(rb)-、-c(ra)=n-、-o-、-si(ra)2-、-s-、-so2-、-n(ra)-和>c=z的基团/原子组成的二价接头基团,其中z选自-o-、-s-和-n(ra)-,并且ra和rb各自独立地选自氢、任选取代的c1-12烷基、任选取代的c2-12烯基、任选取代的c2-12炔基、羟基、任选取代的c1-12烷氧基、c2-12烷氧基烷基、c2-12烯氧基、羧基、c1-12烷氧基羰基、c1-12烷基羰基、甲酰基、芳基、芳氧基-羰基、芳氧基、芳基羰基、杂芳基、杂芳氧基-羰基、杂芳氧基、杂芳基羰基、氨基、单-和二(c1-6烷基)氨基、氨基甲酰基、单-和二(c1-6烷基)-氨基-羰基、氨基-c1-6烷基-氨基羰基、单-和二(c1-6烷基)氨基-c1-6-烷基-氨基羰基、c1-6烷基-羰基氨基、脲基、c1-6-烷酰基氧基、磺基(sulphono)、c1-6烷基磺酰基氧、硝基、叠氮基、硫基(sulphanyl)、c1-6-硫代烷基、卤素、dna嵌入剂、光化学活性基团、热化学活性基团、螯合基团、报告基团和配体,其中芳基和杂芳基可以是任选取代的并且其中两个孪位取代基ra和rb一起可以表示任选取代的亚甲基(=ch2),其中对于所有手性中心,可以找到r或s方向的不对称基团,和;存在的取代基r1*、r2、r3、r5、r5*、r6和r6*中的每个独立地选自氢、任选取代的c1-12烷基、任选取代的c2-12烯基、任选取代的c2-12炔基、羟基、c1-12烷氧基、c2-12烷氧基烷基、c2-12烯氧基、羧基、c1-12烷氧基羰基、c1-12烷基羰基、甲酰基、芳基、芳氧基-羰基、芳氧基、芳基羰基、杂芳基、杂芳氧基-羰基、杂芳氧基、杂芳基羰基、氨基、单-和二(c1-6烷基)氨基、氨基甲酰基、单-和二(c1-6烷基)-氨基-羰基、氨基-c1-6烷基-氨基羰基、单-和二(c1-6烷基)氨基-c1-6-烷基-氨基羰基、c1-6烷基-羰基氨基、脲基、c1-6-烷酰基氧、磺基、c1-6烷基磺酰基氧、硝基、叠氮基、硫基、c1-6-硫代烷基、卤素、dna嵌入剂、光化学活性基团、热化学活性基团、螯合基团、报告基团和配体,其中芳基和杂芳基可以是任选取代的,并且其中两个孪位取代基一起可以表示氧代、硫代、亚氨基,或任选取代的亚甲基;其中rn选自氢和c1-4烷基,并且其中两个邻近的(非孪位)取代基可以表示另外形成双键的键;并且rn*,存在并且不涉及双游离基时,选自氢和c1-4烷基;及其碱盐和酸加成盐。对于所有手性中心,可以找到r或s方向的不对称基团。在一些实施方案中,r4*和r2*一起表示选自c(rarb)-c(rarb)-、c(rarb)-o-、c(rarb)-nra-、c(rarb)-s-和c(rarb)-c(rarb)-o-的双游离基,其中每个ra和rb可以任选独立地选择。在一些实施方案中,ra和rb可以任选独立地选自氢和c1-6烷基,如甲基,如氢。在一些实施方案中,r4*和r2*一起表示双游离基-o-ch(ch2och3)-(2’o-甲氧基乙基双环核酸-seth等,2010,j.org.chem),为-r-或s-构型。在一些实施方案中,r4*和r2*一起表示双游离基-o-ch(ch2ch3)-(2’o-乙基双环核酸-seth等,2010,j.org.chem),为-r-或s-构型。在一些实施方案中,r4*和r2*一起表示双游离基-o-ch(ch3)-,-r-或s-构型。在一些实施方案中,r4*和r2*一起表示双游离基-o-ch2-o-ch2-(seth等,2010,j.org.chem)。在一些实施方案中,r4*和r2*一起表示双游离基-o-nr-ch3-(seth等,2010,j.org.chem)。在一些实施方案中,lna单元具有选自以下组的结构:在一些实施方案中,r1*、r2、r3、r5、r5*独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基。对于所有手性中心,可以找到r或s方向的不对称基团。在一些实施方案中,r1*、r2、r3、r5、r5*是氢。在一些实施方案中,r1*、r2、r3独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基。对于所有手性中心,可以找到r或s方向的不对称基团。在一些实施方案中,r1*、r2和r3是氢。在一些实施方案中,r5和r5*各自独立地选自h、-ch3、-ch2-ch3、-ch2-o-ch3和-ch=ch2。在一些实施方案中,合适地,r5或r5*是氢,其中与其他基团(r5或r5*分别地)一样选自c1-5烷基、c2-6烯基、c2-6炔基、取代的c1-6烷基、取代的c2-6烯基、取代的c2-6炔基或取代的酰基(-c(=o)-);其中每个取代的基团是单或多取代的,取代基独立地选自卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基、取代的c2-6炔基、oj1、sj1、nj1j2、n3、cooj1、cn、o-c(=o)nj1j2、n(h)c(=nh)nj,j2或n(h)c(=x)n(h)j2,其中x是o或s;并且每个j1和j2独立地是h、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基、取代的c2-6炔基、c1-6氨基烷基或取代的c1-6氨基烷基或保护基团。在一些实施方案中,r5或r5*是取代的c1-6烷基。在一些实施方案中,r5或r5*是取代的亚甲基,其中优选的取代基包括一个或多个独立地选自f、nj1j2、n3、cn、oj1、sj1、o-c(=o)nj1j2、n(h)c(=nh)nj,j2或n(h)c(o)n(h)j2的基团。在一些实施方案中,每个j1和j2独立地是h或c1-6烷基。在一些实施方案中,r5或r5*是甲基、乙基或甲氧基甲基。在一些实施方案中,r5或r5*是甲基。在再一个实施方案中,r5或r5*是乙烯基。在一些实施方案中,r5或r5*是取代的酰基。在一些实施方案中,r5或r5*是c(=o)nj1j2。对于所有手性中心,可以找到r或s方向的不对称基团。这样的5’修饰的双环核苷酸公开于wo2007/134181中,将其全部在此并入本文作为参考。在一些实施方案中,b是核碱基,包括核碱基类似物和天然产生的核碱基,如嘌呤或嘧啶,或取代的嘌呤或取代的嘧啶,如本文中提及的核碱基,如选自腺嘌呤、胞嘧啶、胸腺嘧啶、腺嘌呤、尿嘧啶和/或修饰的或取代的核碱基,如5’-噻唑-尿嘧啶、2-硫代-尿嘧啶、5-丙炔基-尿嘧啶、2’硫代-胸腺嘧啶、5-甲基胞嘧啶、5-噻唑-胞嘧啶、5-丙炔基-胞嘧啶和2,6-二氨基嘌呤。在一些实施方案中,r4*和r2*一起表示选自-c(rarb)-o-、-c(rarb)-c(rcrd)-o-、-c(rarb)-c(rcrd)-c(rerf)-o-、-c(rarb)-o-c(rcrd)-、-c(rarb)-o-c(rcrd)-o-、-c(rarb)-c(rcrd)-、-c(rarb)-c(rcrd)-c(rerf)-、-c(ra)=c(rb)-c(rcrd)-、-c(rarb)-n(rc)-、-c(rarb)-c(rcrd)-n(re)-、-c(rarb)-n(rc)-o-和-c(rarb)-s-、-c(rarb)-c(rcrd)-s-的双游离基,其中ra、rb、rc、rd、re和rf各自独立地选自氢、任选取代的c1-12烷基、任选取代的c2-12烯基、任选取代的c2-12炔基、羟基、c1-12烷氧基、c2-12烷氧基烷基、c2-12烯氧基、羧基、c1-12烷氧基羰基、c1-12烷基羰基、甲酰基、芳基、芳氧基-羰基、芳氧基、芳基羰基、杂芳基、杂芳氧基-羰基、杂芳氧基、杂芳基羰基、氨基、单-和二(c1-6烷基)氨基、氨基甲酰基、单-和二(c1-6烷基)-氨基-羰基、氨基-c1-6烷基-氨基羰基、单-和二(c1-6烷基)氨基-c1-6-烷基-氨基羰基、c1-6烷基-羰基氨基、脲基、c1-6-烷酰基氧、磺基、c1-6烷基磺酰基氧、硝基、叠氮基、硫基、c1-6-硫代烷基、卤素、dna嵌入剂、光化学活性基团、热化学活性基团、螯合基团、报告基团和配体,其中芳基和杂芳基可以是任选取代的并且其中两个孪位取代基ra和rb一起可以表示任选取代的亚甲基(=ch2)。对于所有手性中心,可以找到r或s方向的不对称基团。在再一个实施方案中,r4*和r2*一起表示选自-ch2-o-、-ch2-s-、-ch2-nh-、-ch2-n(ch3)-、-ch2-ch2-o-、-ch2-ch(ch3)-、-ch2-ch2-s-、-ch2-ch2-nh-、-ch2-ch2-ch2-、-ch2-ch2-ch2-o-、-ch2-ch2-ch(ch3)-、-ch=ch-ch2-、-ch2-o-ch2-o-、-ch2-nh-o-、-ch2-n(ch3)-o-、-ch2-o-ch2-、-ch(ch3)-o-,和–ch(ch2-o-ch3)-o-,和/或-ch2-ch2-和-ch=ch-的双游离基(二价基团)。对于所有手性中心,可以找到r或s方向的不对称基团。在一些实施方案中,r4*和r2*一起表示双游离基c(rarb)-n(rc)-o-,其中ra和rb独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基,如氢,并且;其中rc选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基,如氢。在一些实施方案中,r4*和r2*一起表示双游离基c(rarb)-o-c(rcrd)-o-,其中ra、rb、rc和rd独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基,如氢。在一些实施方案中,r4*和r2*形成双游离基-ch(z)-o-,其中z选自c1-6烷基、c2-6烯基、c2-6炔基、取代的c1-6烷基、取代的c2-6烯基、取代的c2-6炔基、酰基、取代的酰基、取代的酰胺、硫醇或取代的硫醇;和其中每个取代的基团独立地是单或多取代的,并且任选保护的取代基独立地选自卤素、氧、羟基、oj1、nj1j2、sj1、n3、oc(=x)j1、oc(=x)nj1j2、nj3c(=x)nj1j2和cn,其中每个j1、j2和j3独立地是h或c1-6烷基,并且x是o、s或nj1。在一些实施方案中,z是c1-6烷基或取代的c1-6烷基。在一些实施方案中,z是甲基。在一些实施方案中,z是取代的c1-6烷基。在一些实施方案中,所述取代基是c1-6烷氧基。在一些实施方案中,z是ch3och2-。对于所有手性中心,可以找到r或s方向的不对称基团。这样的双环核苷酸公开于us7,399,845中,将其全部在此并入本文作为参考。在一些实施方案中,r1*、r2、r3、r5、r5*是氢。在一些实施方案中,r1*、r2、r3*是氢,并且r5、r5*中的一个或两个可以是氢以外的基团,如以上和wo2007/134181中所述的。在一些实施方案中,r4*和r2*一起表示在桥中包含取代的氨基基团的双游离基,如由双游离基-ch2-n(rc)-组成或包含双游离基-ch2-n(rc)-,其中rc是c1-12烷氧基。在一些实施方案中,r4*和r2*一起表示双游离基cq3q4-nor-,其中q3和q4独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基;其中每个取代的基团独立地是单或多取代的,并且取代基独立地选自卤素、oj1、sj1、nj1j2、cooj1、cn、o-c(=o)nj1j2、n(h)c(=nh)nj1j2或n(h)c(=x=n(h)j2,其中x是o或s;并且每个j1和j2独立地是h、c1-6烷基,c2-6烯基、c2-6炔基、c1-6氨基烷基或保护基团。对于所有手性中心,可以找到r或s方向的不对称基团。这样的双环核苷酸公开于wo2008/150729中,将其全部在此并入本文作为参考。在一些实施方案中,r1*、r2、r3、r5、r5*独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基。在一些实施方案中,r1*、r2、r3、r5、r5*是氢。在一些实施方案中,r1*、r2、r3是氢,并且r5、r5*中的一个或两个可以是氢以外的基团,如以上和wo2007/134181中所述的。在一些实施方案中,r4*和r2*一起表示双游离基(二价基团)c(rarb)-o-,其中ra和rb各自独立地是氢、c1-c12烷基、取代的c1-c12烷基、c2-c12烯基、取代的c2-c12烯基、c2-c12炔基、取代的c2-c12炔基、c1-c12烷氧基、取代的c1-c12烷氧基、oj1sj1、soj1、so2j1、nj1j2、n3、cn、c(=o)oj1、c(=o)nj1j2、c(=o)j1、o-c(=o)nj1j2、n(h)c(=nh)nj1j2、n(h)c(=o)nj1j2或n(h)c(=s)nj1j2;或ra和rb一起是=c(q3)(q4);q3和q4各自独立地是氢、卤素、c1-c12烷基或取代的c1-c12烷基;每个取代的基团独立地是单或多取代的,取代基独立地选自卤素、c1-c6烷基、取代的c1-c6烷基、c2-c6烯基、取代的c2-c6烯基、c2-c6炔基、取代的c2-c6炔基、oj1、sj1、nj1j2、n3、cn,c(=o)oj1、c(=o)nj1j2、c(=o)j1、o-c(=o)nj1j2、n(h)c(=o)nj1j2或n(h)c(=s)nj1j2,并且每个j1和j2独立地是h、c1-c6烷基、取代的c1-c6烷基、c2-c6烯基、取代的c2-c6烯基、c2-c6炔基、取代的c2-c6炔基、c1-c6氨基烷基、取代的c1-c6氨基烷基或保护基团。这样的化合物公开于wo2009006478a,将其全部在此并入本文作为参考。在一些实施方案中,r4*和r2*形成双游离基-q-,其中q是c(q1)(q2)c(q3)(q4)、c(q1)=c(q3)、c[=c(q1)(q2)]-c(q3)(q4)或c(q1)(q2)-c[=c(q3)(q4)];q1、q2、q3、q4各自独立地是h、卤素、c1-12烷基、取代的c1-12烷基、c2-12烯基、取代的c1-12烷氧基、oj1、sj1、soj1、so2j1、nj1j2、n3、cn、c(=o)oj1、c(=o)-nj1j2、c(=o)j1、-c(=o)nj1j2、n(h)c(=nh)nj1j2、n(h)c(=o)nj1j2或n(h)c(=s)nj1j2;每个j1和j2独立地是h、c1-6烷基、c2-6烯基、c2-6炔基、c1-6氨基烷基或保护基团;并且,任选其中q是c(q1)(q2)c(q3)(q4)且q3或q4中的一个是ch3时,那么其他q3或q4中的至少一个或q1和q2中的一个是氢以外的基团。在一些实施方案中,r1*、r2、r3、r5、r5*是氢。对于所有手性中心,可以找到r或s方向的不对称基团。这样的双环核苷酸公开于wo2008/154401中,将其全部在此并入本文作为参考。在一些实施方案中,r1*、r2、r3、r5、r5*独立地选自氢、卤素、c1-6烷基、取代的c1-6烷基、c2-6烯基、取代的c2-6烯基、c2-6炔基或取代的c2-6炔基、c1-6烷氧基、取代的c1-6烷氧基、酰基、取代的酰基、c1-6氨基烷基或取代的c1-6氨基烷基。在一些实施方案中,r1*、r2、r3、r5、r5*是氢。在一些实施方案中,r1*、r2、r3是氢,并且r5、r5*中的一个或两个可以是氢以外的基团,如以上和wo2007/134181或wo2009/067647(α-l-双环核酸类似物)中所述的。更多双环核苷类似物及其在反义寡核苷酸中的用途公开于wo2011115818、wo2011/085102、wo2011/017521、wo09100320、wo10036698、wo09124295&wo09006478。在一些方面中,这样的核苷类似物在本发明的化合物中是有用的。在一些实施方案中,本发明的寡核苷酸化合物中使用的lna优选具有通式ii的结构:其中y选自-o-、-ch2o-、-s-、-nh-、n(re)和/或-ch2-;z和z*独立地选自核苷酸间键、rh、端基或保护基团;b构成天然或非天然核苷酸碱基部分(核碱基),并且rh选自氢和c1-4烷基;ra、rb、rc、rd和re任选独立地选自氢、任选取代的c1-12烷基、任选取代的c2-12烯基、任选取代的c2-12炔基、羟基、c1-12烷氧基、c2-12烷氧基烷基、c2-12烯氧基、羧基、c1-12烷氧基羰基、c1-12烷基羰基、甲酰基、芳基、芳氧基-羰基、芳氧基、芳基羰基、杂芳基、杂芳氧基-羰基、杂芳氧基、杂芳基羰基、氨基、单-和二(c1-6烷基)氨基、氨基甲酰基、单-和二(c1-6烷基)-氨基-羰基、氨基-c1-6烷基-氨基羰基、单-和二(c1-6烷基)氨基-c1-6-烷基-氨基羰基、c1-6烷基-羰基氨基、脲基、c1-6-烷酰基氧、磺基、c1-6烷基磺酰基氧、硝基、叠氮基、硫基、c1-6-硫代烷基、卤素、dna嵌入剂、光化学活性基团、热化学活性基团、螯合基团、报告基团和配体,其中芳基和杂芳基可以是任选取代的并且其中两个孪位取代基ra和rb一起可以表示任选取代的亚甲基(=ch2);并且rh选自氢和c1-4烷基。在一些实施方案中,ra、rb、rc、rd和re任选独立地选自氢和c1-6烷基,如甲基。对于所有手性中心,可以找到r或s方向的不对称基团,例如,两个示例性立体化学异构体包括β-d和α-l-异构体,其可以是如下所示:以下显示了特定的示例性lna单元:术语“硫代-lna”包含其中以上通式中的y选自s或-ch2-s-的锁核苷酸。硫代-lna可以是β-d和α-l-构型。术语“氨基-lna”包含其中以上通式中的y选自-n(h)-、n(r)-、ch2-n(h)-和-ch2-n(r)-的锁核苷酸,其中r选自氢和c1-4烷基。氨基-lna可以是β-d和α-l-构型。术语“氧基-lna”包含其中以上通式中的y表示-o-的锁核苷酸。氧基-lna可以是β-d和α-l-构型。术语“ena”包含其中以上通式中的y是-ch2-o-的锁核苷酸(其中-ch2-o-的氧原子连接相对于碱基b的2’-位置)。re是氢或甲基。在一些示例性实施方案中,lna选自β-d-氧基-lna、α-l-氧基-lna、β-d-氨基-lna和β-d-硫代-lna,特别是β-d-氧基-lna。方案1中展示lna核苷的某些例子。方案1如实例中所示,在本发明的一些实施方案中,寡核苷酸中的lna核苷是β-d-氧基-lna核苷。缺口聚物如本文中使用的术语“缺口聚物”是指包含募集rnaseh的寡核苷酸区域(缺口)的反义寡核苷酸,所述寡核苷酸区域的5’和3’两侧为包含一个或多个增强亲和性的修饰核苷的区域(侧翼或翼)。本文中描述了多种缺口聚物设计。头聚物和尾聚物是能够募集rnaseh的寡核苷酸,其中侧翼中的一个缺失,即,只有一个寡核苷酸的末端包含增强亲和性的修饰核苷。对于头聚物,3’侧翼缺失(即,5’侧翼包含增强亲和性的修饰核苷),以及对于尾聚物,5’侧翼缺失(即,3’侧翼包含增强亲和性的修饰核苷)。缺口聚物设计缺口聚物寡核苷酸广泛用于通过反义机制抑制细胞中的靶rna,如mrna或病毒rna(并且因此也可以称为反义缺口聚物寡核苷酸)。在缺口聚物结构中,寡核苷酸包含至少三个不同的结构区5’-侧翼、缺口和3’-侧翼,5’->3’方向的f-g-f’。g区或缺口区包含至少5个连续核苷酸的区域,其能够募集rnaseh,如dna核苷酸的区域,例如,6-14个dna核苷酸或能够募集rnaseh的其他核苷,例如,α-l-氧基-lna、2’-氟-ana和una。缺口区5’和3’两侧为包含一个或多个增强亲和性的修饰核苷的区域(f和f’也称为侧翼区或翼区),如2’修饰的核苷酸。在一些实施方案中,侧翼区可以是1-8个核苷酸长。在更多实施方案中,区域f(5’侧翼或5’翼)连接至区域g的5’端,并且区域f’(3’侧翼或3’翼)连接区域g的’3端。区域f和f’独立地包含至少一个修饰的核苷、独立地含有至少一个修饰的核苷或独立地由至少一个修饰的核苷组成,如至少2个、至少3个、至少4个、至少5个、至少6个、至少7个修饰的核苷。在一个实施方案中,区域f和/或f’独立地包含1至7个修饰的核苷或独立地由1-7个修饰的核苷组成,如2至6个修饰的核苷,如2至5个修饰的核苷,如2至4个修饰的核苷,如1至3个修饰的核苷,如1、2、3或4个修饰的核苷。通过在区域的5’端和3’端具有至少一个修饰的核苷来定义f区。通过在区域的5’端和3’端具有至少一个修饰的核苷来定义f’区。在一些实施方案中,区域f和/或f’中的修饰的核苷具有3’桥(endo)结构。在一个实施方案中,区域f和/或f’中的一个或多个修饰的核苷是2’修饰的核苷。在一个实施方案中,区域f和/或f’中的所有核苷是2’修饰的核苷。在另一个实施方案中,除了2’修饰的核苷,区域f和/或f’包含dna和/或rna。包含dna和/或rna的侧翼的特征在于在f和/或f’区的5’端和3’端具有2’修饰的核苷。在一个实施方案中,区域f和/或f’包含dna核苷,如1至3个连续的dna核苷,如1至3个或1至2个连续的dna核苷。侧翼中的dna核苷优选不能够募集rnaseh。在一些实施方案中,f和/或f’区域中的2’修饰的核苷和dna和/或rna与1至3个2’修饰的核苷和1至3个dna和/或rna核苷交替。这样的侧翼也可以称为交替侧翼。具有交替侧翼的寡核苷酸中的区域f和/或f’的长度可以独立地为4至10个核苷,如4至8个,如4至6个核苷,如4、5、6或7个修饰的核苷。在一些实施方案中,只有寡核苷酸的f区是交替的。在一些实施方案中,只有寡核苷酸的f’区是交替的。具有交替核苷的区域f和/或f’的特定实例为2’1-3-n’1-4-2’1-32’1-2-n’1-2-2’1-2-n’1-2-2’1-2其中2’表示修饰的核苷,并且n’是rna或dna。在一些实施方案中,交替侧翼中的所有修饰的核苷是lna,并且n’是dna。在再一个实施方案中,区域f和/或f’中的一个或多个2’修饰的核苷独立地选自2’-o-烷基-rna单元、2’-o-甲基-rna、2’-氨基-dna单元、2’-氟-dna单元、2’-烷氧基-rna、moe单元、lna单元、阿糖核酸(ana)单元和2’-氟-ana-单元。在一些实施方案中,f和/或f’区包含lna和2’取代的修饰的核苷。常常将这些称为混合翼或混合侧翼寡核苷酸。在本发明的一个实施方案中,区域f和/f’中的所有修饰的核苷是lna核苷。在再一个实施方案中,区域f和/或f’中的所有核苷是lna核苷。在再一个实施方案中,区域f和/或f’中的lna核苷独立地选自氧基-lna、硫代-lna、氨基-lna、cet和/或ena,以β-d或α-l构型或其组合。在优选实施方案中,区域f和/或f’在连续序列的5’端包含至少1个β-d-氧基lna单元。在进一步的实施方案中,区域g(缺口区)优选包含至少4个能够募集上述核酸酶(特别是rnaseh)的连续核苷、含有至少4个能够募集上述核酸酶(特别是rnaseh)的连续核苷或由至少4个能够募集上述核酸酶(特别是rnaseh)的连续核苷组成,如至少5个,如至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个或至少16个。在再一个实施方案中,区域g包含5至12个能够募集上述核酸酶的连续核苷酸单元,或由5至12个能够募集上述核酸酶的连续核苷酸单元组成,或6至10个或7至9个,如8个。在一个实施方案中,区域g中能够募集核酸酶的核苷选自dna、α-l-lna、c4’烷基化dna(如pct/ep2009/050349和vester等,bioorg.med.chem.lett.18(2008)2296-2300中所述的,在此并入本文作为参考)、阿拉伯糖衍生的核苷如ana和2’f-ana(mangos等,2003j.am.chem.soc.125,654-661)、una(未锁核酸)(如描述于fluiter等,mol.biosyst.,2009,10,1039,在此并入本文作为参考)。una是未锁核苷,通常其中核糖的c2和c3之间的键已经去除,形成未锁的“糖”残基。在再一个实施方案中,区域g中的至少一个核苷单元是dna核苷单元,如1至12个dna单元,如2、3、4、5、6、7、8、9、10或11个dna单元,优选2至12个dna单元,如4至12个dna单元,更优选5至11个,或2至10个,4至10个或6至10个dna单元,如7至10个dna单元,最优选8、9或10个dna单元。在一些实施方案中,区域g由100%dna单元组成。在再一个实施方案中,区域g可以由能够介导rnaseh切割的dna和其他核苷的混合物组成。区域g可以由至少50%dna,更优选60%、70%或80%dna组成,并且甚至更优选90%或95%dna。在再一个实施方案中,区域g中的至少一个核苷单元是α-l-lna核苷单元,如至少一个α-l-lna,如2、3、4、5、6、7、8或9个α-l-lna。在再一个实施方案中,区域g包含至少一个α-l-lna是α-l-氧基-lna。在再一个实施方案中,区域g包含dna和α-l-lna核苷单元的组合。在一些实施方案中,区域g中的连续序列的大小可以较长,如12、13、14、15、16、17、18、19或20个核苷单元。在一些实施方案中,区域g中的核苷具有2’桥结构。在本文中记载的缺口聚物设计中,缺口区(y’)可以包含一个或多个立体限定的硫代磷酸酯键,并且缺口区剩余的核苷间键可以例如是非立体限定的核苷间键,或也可以是立体限定的硫代磷酸酯键。lna缺口聚物术语lna缺口聚物是其中至少一个增强亲和性的修饰核苷是lna核苷的缺口聚物寡核苷酸。在一些实施方案中,缺口聚物寡核苷酸的两个侧翼都包含至少一个lna单元,并且在一些实施方案中,侧翼的全部核苷是lna核苷。在一些实施方案中,3’侧翼包含至少一个lna核苷,优选至少2个lna核苷。在一些实施方案中,5’侧翼包含至少一个lna核苷。在一些实施方案中,5’和3’侧翼区包含lna核苷。在一些实施方案中,侧翼区中的所有核苷是lna核苷。通常,lna缺口聚物的侧翼的lna载量低于2’取代的核苷,并且lna缺口聚物设计的实例包括[lna]1-4-[dna]5-15-[lna]1-4。在一些实施方案中,缺口聚物是所谓的短聚物,如wo2008/113832中所述的,在此并入本文作为参考。更多缺口聚物设计公开于wo2004/046160、wo2007/146511中,在此并入本文作为参考。混合翼缺口聚物术语混合翼缺口聚物或混合侧翼缺口聚物是指其中至少一个侧翼区包含至少一个lna核苷和至少一个非lna修饰的核苷的lna缺口聚物,所述非lna修饰的核苷如至少一个dna核苷或至少一个2’取代的修饰核苷,如,例如,2’-o-烷基-rna、2’-o-甲基-rna、2’-烷氧基-rna、2’-o-甲氧基乙基-rna(moe)、2’-氨基-dna、2’-氟-rna和2’-f-ana核苷。在一些实施方案中,混合翼缺口聚物具有一个只包含lna核苷的侧翼(例如,5’或3’),而另一个侧翼(相对的3’或5’)包含2’取代的修饰核苷和任选的lna核苷。缺口打破者(gapbreaker)与能够维持rnaseh募集的缺口聚物相关地使用术语“缺口打破者寡核苷酸”,即使通过非rnaseh募集核苷(缺口打破者核苷,e)破坏缺口区,使得缺口区包含少于5个连续的dna核苷。非rnaseh募集核苷例如是3’桥构象的核苷,如其中核苷的核糖糖环的c2’和c4’之间的桥是β构象的lna,如β-d-氧基lna或scet核苷。缺口打破者寡核苷酸募集rnaseh的能力通常是序列或甚至化合物特异性的-参见rukov等,2015nucl.acidsres.vol.43pp.8476-8487,其公开了募集rnaseh的“缺口打破者”寡核苷酸,这在一些情况下提供了更特异性的靶rna的切割。在一些实施方案中,本发明的寡核苷酸是缺口打破者寡核苷酸。在一些实施方案中,缺口打破者寡核苷酸包含5’-侧翼(f)、缺口(g)和3’-侧翼(f’),其中缺口被非rnaseh募集核苷(缺口打破者核苷,e)破坏,使得缺口含有至少3或4个连续的dna核苷。缺口打破者设计是基于缺口聚物设计,例如,本文中公开的那些(例如,区域f对应于以上缺口聚物的x’区,而区域f’对应于本文中所述的缺口聚物的区域z’),除了缺口区(区域y’)包含缺口打破者核苷。在一些实施方案中,缺口打破者核苷(e)是lna核苷,其中核苷的核糖糖环的c2’和c4’之间的桥是β构象的,并且放置在缺口区内,使得缺口打破者lna核苷的5’和3’侧为至少3个(5’)和3个(3’)或至少3个(5’)和4个(3’)或至少4个(5’)和3个(3’)dna核苷,并且其中寡核苷酸能够募集rnaseh。缺口打破者寡核苷酸通过下式来表示:f-g-e-g-f’;特别是f1-7-g3-4-e1-g3-4-f’1-7d’-f-g-f’,特别是d’1-3-f1-7-g3-4-e1-g3-4-f’1-7f-g-f’-d”,特别是f1-7-g3-4-e1-g3-4-f’1-7-d”1-3d’-f-g-f’-d”,特别是d’1-3-f1-7-g3-4-e1-g3-4-f’1-7-d”1-3其中g表示dna核苷,并且区域d’和d”是任选的,并且可以是另外的5’和/或3’核苷,如dna核苷。在一些实施方案中,缺口打破者核苷(e)是lna、β-d-氧基lna或scet或另一个lna核苷,如本文中公开的β-d-核苷。立体限定的缺口聚物在一些实施方案中,源自亲本缺口聚物寡核苷酸的子寡核苷酸具有至少一个是立体限定的缺口区的核苷间键,并且任选其中缺口区包含rp和sp核苷间键。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,缺口区的至少一个核苷间键是立体限定的,并且其中中心区包含rp和sp核苷间键。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,区域g内的核苷间键是全部立体限定的硫代磷酸酯核苷间键。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,区域f和f’内的核苷间键是立体限定的硫代磷酸酯核苷间键。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,区域f和g之间以及区域g和f’之间的核苷间键是立体限定的硫代磷酸酯核苷间键。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,区域f-g-f’的连续核苷内的所有核苷间键是立体限定的硫代磷酸酯核苷间键。在寡核苷酸的缺口区中引入至少一个立体限定的硫代磷酸酯键可以用于调节寡核苷酸的生物特征,例如,可以调节毒性特征。在一些实施方案中,子寡核苷酸(和任选地亲本)中的缺口区中的2、3、4或5个硫代磷酸酯键是立体限定的。在一些实施方案中,缺口区中的剩余核苷间键不是立体限定的。它们作为寡核苷酸物质群中的rp和sp的外消旋混合物存在。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,寡核苷酸中的剩余核苷间键不是立体限定的。在一些实施方案中,在子寡核苷酸(和任选地亲本)中,缺口区中的所有核苷间键是立体限定的。在一些实施方案中,寡聚物的缺口区中的1、2、3、4、5、6、7、8、9、10、11、12、13或14个键是立体选择性的硫代磷酸酯键。在一些实施方案中,寡聚物(例如,缺口聚物)中的5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%的键是立体选择性的硫代磷酸酯键。在一些实施方案中,寡聚物中的所有硫代磷酸酯键是立体选择性的硫代磷酸酯键。在一些实施方案中,寡聚物中的所有核苷间键是立体限定的硫代磷酸酯键。rnaseh活性和募集反义寡核苷酸的rnaseh活性是指其在与互补rna分子的双链体中时募集rnaseh的能力。wo01/23613提供了用于测定rnaseh活性的体外方法,其可以用于测定募集rnaseh的能力。通常,如果与互补靶核酸序列一起提供时,具有使用与待测试的修饰寡核苷酸具有相同碱基序列但只含有在寡核苷酸中的所有单体之间具有硫代磷酸酯键的dna单体的寡核苷酸和使用wo01/23613(在此并入本文作为参考)的实施例91-95提供的方法时测定的初始速率的至少5%,如至少10%或高于20%的初始速率(以pmol/l/min表示)时,则认为寡核苷酸能够募集rnaseh。功效和治疗指数除了毒性之外,还可以作为本发明方法的一部分测试通过本发明的方法鉴定的核酸分子,以确定它们仍然是有效的核酸分子。特别地,使用基于核酸的药物,已经观察到降低毒性时,一部分是由于药物的功效降低引起的。在本发明的一个方面中,毒性等级的降低没有导致功效的显著降低。在一些实施方案中,与毒性参照物质相比时,根据本发明的方法选择的药物的治疗指数(ti)提高了。为了本发明的目的,ti是指引起50%受试者的肾脏中不良作用的药物的剂量(td50)除以导致50%受试者中所需功效(靶敲低或药理学作用)的剂量(ec50)。在一些实施方案中,与毒性参照药物(如本文中的化合物4-1或有毒亲本寡核苷酸或有毒亲本药物)相比时,ti提高至少10%,如至少15、25、50、75、100、150、200、250或300%。为了实验目的,认为靶存在于小鼠中,可以在七天小鼠研究中测定td50和ec50。如果例如对于核酸分子,小鼠中的序列保守是不利的,则可以使用其他模式物种,例如,大鼠、猴子、狗、猪或猴子(例如,食蟹猴)。在一些实施方案中,ti提高是由于提高的td50和ed50的低或无降低引起的。ti提高也可以是两个参数改善的结果。关于本发明的方法,ti的提高不仅仅是通过提高药物的功效(即,降低的ec50)引起的。本发明的方法因此可以包括额外的针对调节(例如,抑制)靶的功效筛选(例如,子)药物的步骤。备选地,本发明的方法可以包括另外的测试选定的具有降低毒性的药物(例如,立体限定的寡核苷酸变体)的步骤,以测定其作为药物(例如,作为反义寡核苷酸)的功效。对于反义寡核苷酸,可以通过寡核苷酸募集rnaseh的能力来确定功效,或在一些实施方案中,可以是在体外调节细胞中的靶表达的能力,或在一些实施方案中,在体内。缀合物部分在一些实施方案中,缀合物部分包含或是碳水化合物、非核苷糖、碳水化合物复合物。在一些实施方案中,碳水化合物选自半乳糖、乳糖、n-乙酰基半乳糖胺、甘露糖和甘露糖-6-磷酸。在一些实施方案中,缀合物部分包含或选自蛋白质、糖蛋白、多肽、肽、抗体、酶和抗体片段。在一些实施方案中,缀合物部分是亲脂性部分,如选自脂质、磷脂、脂肪酸和甾醇的部分。在一些实施方案中,缀合物部分选自小分子药物、毒素、报告分子和受体配体。在一些实施方案中,缀合物部分是聚合物,如聚乙二醇(peg)、聚丙二醇。在一些实施方案中,缀合物部分是或包含去唾液酸糖蛋白受体靶向部分,其可以包括,例如,半乳糖、半乳糖胺、n-甲酰基-半乳糖胺、n-乙酰基半乳糖胺、n-丙酰基-半乳糖胺、n-正丁酰基-半乳糖胺和n-异丁酰基半乳糖胺。在一些实施方案中,缀合物部分包含半乳糖簇(cluster)例如n-乙酰半乳糖胺三聚体。在一些实施方案中,缀合物部分包含galnac(n-乙酰基半乳糖胺),如单价、二价、三价或四价galnac。三价galnac缀合物可以用于将化合物靶向肝(参见,例如,us5,994517和hangeland等,bioconjugchem.1995年11-12;6(6):695-701,wo2009/126933、wo2012/089352、wo2012/083046、wo2014/118267、wo2014/179620&wo2014/179445),参见图2中的特定实例。将其中使用的这些galnac参照和特定的缀合物在此并入本文作为参考。发明详述体内毒性的预测本文中所述的方法可以用于预测药物在哺乳动物中的体内毒性。任何化合物的毒性通常取决于其剂量,并且因此本发明的方法可以用于评价化合物比较毒性特征,所述特征与阴性对照和/或其毒性特征已知的药物相比,或与核酸分子的群(或文库)相比,如核酸分子的文库。在这个方面中,体内毒性的预测可以是将药物在哺乳动物中体内给药时,遇到毒性表型(如肾毒性)的比较风险的评价。评价药物的安全性时,研究的参数之一是物质对肾脏的影响,例如,在肾近端小管中的累积、肾小管退化/恢复变化(肾小管毒性(tubulotoxicity))、针对急性肾损伤的生物标志物,如kim-1,其在尿液中是可检测的。肾相关的毒性通常称为肾毒性。本发明的方法可以用于预测药物在哺乳动物体内的体内毒性(例如,肾毒性),或者换句话说用于测定药物在哺乳动物体内的体内毒性特征的可能性(例如,肾毒性)。在本发明的一个实施方案中,体内肾毒性是急性肾损伤和/或肾小管退化。本发明的方法可以用于鉴定以调节靶有效的剂量或治疗有效剂量使用时在体内无毒的(例如,不是肾毒性的)药物。本发明的方法因此允许选择以有效剂量在体内使用时没有呈现剂量限制性毒性(例如,肾毒性)的药物。将认识到选择用于体内或用于治疗用途的药物时具有宽的安全边际是有利的,并且因此本发明的方法可以用于鉴定或选择以有效或较高剂量(例如,高达2×有效剂量,或高达3×有效剂量,或高达5×有效剂量,或高达10×有效剂量)给药时没有引起体内毒性(如,肾毒性)的药物。本发明的方法因此可以用于鉴定用于体内使用的具有高于(例如,至少2×,至少3×,至少5×,至少10×)有效剂量的最大耐受剂量的药物。在这点上,本发明的方法可以用于选择对于安全治疗给药具有合适的治疗指数(ti)或如本文中定义的提高的治疗指数的药物。本发明确定了egf作为体外基于细胞的测定法中的可靠生物标志物,用于预测药物的体内肾毒性。本发明的一个方面是用于预测药物在哺乳动物中的体内肾毒性的体外方法,所述方法包括步骤:a)在含有至少4ng/ml表皮生长因子(egf)的合适的细胞培养基中培养表达表皮生长因子受体(egfr)的细胞;b)将药物给予所述细胞培养物;c)孵育细胞一段时间;和d)随后测量上清液中的egf水平;其中上清液中的egf的提高表示药物是或预测是与肾毒性相关。可以相对于获自未处理的细胞、用溶媒处理的细胞(溶媒对照,即,其中溶解药物的溶剂,例如,盐水、dmso或其他合适的溶剂)或用无毒参照药物处理的细胞(其中无毒的药物已经证实了在体内是无毒的(体内证实的无毒药物))的参照值(对照)来评价胞外egf水平的提高。这样的无毒药物可以例如是具有seqidno:1的反义寡核苷酸化合物1-1,特别是如果实施本发明测定法的药物是如反义寡核苷酸的核酸分子。在一些实施方案中,作为参照值的百分比来测定egf生物标志物的改变。如实施例中所示的,本发明的发明人已经发现了上清液中的egf水平(胞外egf)的提高相对于参照值高175%,如至少200%,如至少250%,如至少300%,如至少350%,如至少400%,是药物化合物的肾毒性的预测。在其中药物是反义寡核苷酸的实施方案中,将寡核苷酸以高于10微摩尔,如20至150微摩尔,如30至120微摩尔,如50至100微摩尔的浓度给予细胞。在更多实施方案中,进一步将上清液中的egf水平与获自用肾毒性药物处理的细胞的第二参照值进行比较,其中肾毒性参照药物已经证实了能在体内引起肾毒性(体内证实的肾毒性参照药物)。在一些实施方案中,本发明的方法因此可以进一步包括使用一种或多种具有已知毒性(例如,肾毒性)特征的药物的给药的本发明的方法步骤,所述具有已知毒性特征的药物如已知引发体内肾毒性的阳性对照药物和/或已知不引发体内肾毒性的阴性对照药物,并将来自核酸分子给药的上清液中的egf水平与获自阳性和/或阴性对照的水平进行比较。从阴性对照(溶媒对照或体内证实的无毒药物)和阳性对照(体内证实的肾毒性药物)产生的数据允许确定可以用于针对测定法中批次与批次间的变化归一化的测定窗口。所述测定窗口(aw)是无毒参照药物或溶媒对照和肾毒性参照药物之间的差异。为了比较不同的药物,例如,来自药物文库的药物,测定每种药物的毒性等级是有利的。这根据下式来进行毒性等级可以用于区分药物的肾毒性的水平,或肾毒性的预测水平。在一些实施方案中,毒性等级可以如下用于区分预测的体内肾毒性:预测的体内毒性毒性等级高50-100中等20-50低6-20无害的0-5特别地,毒性等级高于6,如高于20,如高于50时,药物预测为肾毒性的。在一个实施方案中,使用“材料和方法”部分中所述的寡核苷酸处理的条件测定了毒性等级,其中将寡核苷酸以高于10微摩尔(如20至150微摩尔,如30至120微摩尔,如50至100微摩尔)的浓度给予细胞。在一些实施方案中,体内证实的肾毒性参照药物是具有seqidno:4的反义寡核苷酸化合物4-1。额外地或备选地,可以通过使用本发明的方法系列地或平行地比较药物文库(特别是核酸分子文库,如反义寡核苷酸、rnai剂或适体的核酸分子文库),并且比较给予药物文库的每个成员后的egf生物标志物的水平,来确定对照数据。这样的方法可以选择相比较毒性(如肾毒性)较低的药物。在一些实施方案中,对照数据可以是或可以包括来自没有给予药物或溶媒对照的细胞培养物样品的对照数据(0天对照)。这样的样品可以就在给予步骤前立即获得的。补充生物标志物如实施例中所示的,本发明的发明人已经发现了胞内三磷酸腺苷(atp)水平和胞外肾损伤分子-1(kim-1)蛋白水平或胞内kim-1mrna水平可以作为egf生物标志物的补充生物标志物,进一步加强本发明方法的可预测性。在本发明的一个实施方案中,用于预测药物在哺乳动物中的体内肾毒性的方法,所述方法包括步骤:a)在含有至少4ng/ml表皮生长因子(egf)的合适的细胞培养基中培养表达表皮生长因子受体(egfr)的细胞;b)将药物给予所述细胞培养物;c)孵育细胞一段时间;和d)随后测量上清液中的egf水平和胞内atp水平;其中上清液中的egf水平的提高和胞内apt水平的降低表示药物是或预测是与肾毒性相关。相对于参照值(阴性对照)而言,细胞atp降低至相对于参照值的至少80%,如至少约70%,如至少约60%,如至少50%,如至少40%表示增强的引发体内肾毒性的倾向。将atp生物标志物与一组已经相对于egf生物标志物描述的对照进行比较。特别地,与获自未处理的细胞、用溶媒处理的细胞(溶媒对照,即,其中溶解核酸分子的溶剂,例如,盐水、dmso或其他合适的溶剂)或用无毒的参照药物处理的细胞(其中无毒的药物已经证实了在体内是无毒的(体内证实的无毒药物))的参照值进行比较。这样的待实施本发明测定法的无毒药物例如是具有seqidno:1的化合物1-1,特别地如果待实施本发明测定法的药物是核酸分子,如反义寡核苷酸。胞内atp生物标志物可以进一步与获自用肾毒性药物处理的细胞的第二参照值进行比较,其中肾毒性参照药物已经证实在体内引起肾毒性(体内证实的肾毒性参照药物)。在一些实例中,单独的egf生物标志物不足以预测具有已知体内肾毒性的化合物的肾毒性。在这些情况中,kim-1作为体外生物标志物显示出能很好地补充egf生物标志物。如图2中所示的,本发明测定法中egf和kim-1生物标志物的组合导致具有已知体内肾毒性的化合物的100%预测。在本发明的一个实施方案中,用于预测药物在哺乳动物中的体内肾毒性的方法,所述方法包括步骤:a)在含有至少4ng/ml表皮生长因子(egf)的合适的细胞培养基中培养表达表皮生长因子受体(egfr)的细胞;b)将药物给予所述细胞培养物;c)孵育细胞一段时间;和d)随后测量上清液中的egf水平和kim-1蛋白水平;其中上清液中的egf或kim-1水平的提高表示药物是或预测是与肾毒性相关。作为胞外kim-1蛋白水平的备选或补充,还可以测量kim-1mrna水平的胞内水平。在一些实施方案中,作为对照参照值(阴性对照)的百分比来测定胞外kim-1生物标志物的改变。如实施例中所示的,本发明的发明人已经发现了相对于参照值而言,上清液中的kim-1水平(胞外kim-1)的提高高于175%,如至少200%,如至少250%,如至少300%,如至少350%,如至少400%,是药物化合物的肾毒性的预测。特别地,药物是核酸分子时,如反义寡核苷酸,kim-1水平的提高是预测性的,即使对于相同的化合物,没有可观察的egf水平提高和/或atp水平降低。不希望受到理论的束缚,我们认为引发体外kim-1生物标志物提高的机制不同于引发胞外egf水平提高或胞内atp水平降低的机制。应当认识到生物标志物实际的提高/降低水平将取决于许多因素,包括细胞培养物的性质、细胞培养物的密度、所用的药物的浓度和药物的孵育时间。相关参数描述于以下部分中。细胞培养物在本发明的实施例中,已经测试了几种细胞培养物。原代细胞培养物以及无限增殖化细胞培养物显示出在本发明的方法中能起到作用,只要它们表达表皮生长因子受体(egfr)。egfr是内源性的,表明其通过细胞表达。本发明的方法可以用于测定药物在模式物种(如啮齿动物物种,如小鼠、大鼠或兔子)或猪(例如,迷你猪)或狗,或灵长类,如猴子(例如,食蟹猴))中的体内毒性的可能性,或可以用于测定药物在人体内的毒性可能性。发明人已经发现了使用原代egfr表达上皮细胞或原代肝细胞或无限增殖化egfr表达上皮细胞能预测啮齿动物研究以及人临床试验中体内看到的毒性特征。特别地,表达功能性egfr的源自肾脏或肺组织的上皮细胞在本发明的方法中是合适的。在一个实施方案中,上皮细胞不是源自眼睛,特别地不是源自视网膜色素上皮,特别地细胞培养物不是arpe19细胞培养物。在本发明的一个实施方案中,细胞表达功能性egfr,在正常条件(没有接受药物或溶媒物质)下培养时,所述细胞消耗生长培养基中存在的egf。细胞在没有接受药物或溶媒物质的常规条件下生长时,egf的消耗速率优选为72小时内至少50%的来自培养基的egf。一些细胞培养物可以具有较高的egf消耗速率,如60小时内至少50%的培养基中的egf,如48小时内至少50%的培养基中的egf。为了测量细胞培养物中的egf消耗,培养基包含egf是重要的。可以根据实施例8的原理来评价细胞培养物的egf消耗。评价未处理细胞培养物的egf消耗以及测量药物处理后egf水平提高的培养基中需要的egf水平可以随着细胞类型而改变。在一些实施方案中,细胞培养基中的egf包含3至20ngegf/ml培养基,如5至15ng/ml,如6至10ng/ml,如至少3ng/ml,如至少4ng/ml,如至少5ng/ml,如至少6ng/ml,如至少7ng/ml,如至少8ng/ml,如至少9ng/ml。在优选实施方案中,细胞培养基包含8至15ng/ml。在更优选的实施方案中,细胞培养基包含至少10ngegf/ml培养基。在一个实施方案中,表达功能性egfr的细胞可以选自上皮细胞、内皮细胞、间充质细胞、神经外胚层细胞和肝细胞。特别地,将源自上皮细胞或肝细胞的细胞培养物用于本发明的方法中。在一个实施方案中,上皮细胞可以是哺乳动物原代细胞培养物的形式,如选自啮齿动物原代上皮细胞如小鼠或大鼠原代上皮细胞的细胞培养物;猪(例如,迷你猪)上皮细胞、狗上皮细胞和非人灵长类原代上皮细胞,如猴子(例如,食蟹猴)或人原代上皮细胞的细胞培养物。在优选实施方案中,细胞培养物获自大鼠或人原代上皮细胞或肝细胞。除了可获自各种物种,细胞培养物也可以获自各种器官,如来自胃肠道、肺、生殖道和尿道、肾脏、外分泌和内分泌腺的上皮细胞。在一个实施方案中,上皮细胞培养物是肾上皮细胞培养物或肺上皮细胞培养物。由于本发明着手预测肾毒性,因此选自近端小管上皮细胞、远端小管上皮细胞和集合管上皮细胞的肾上皮细胞是特别相关的。原代细胞培养物的特定实例是从人ptec或大鼠ptec细胞制得的那些。除了上皮细胞培养物,本发明的实例还显示出可以使用其他表达egfr的细胞类型,例如,原代肝细胞。在另一个实施方案中,表达egfr的细胞培养自无限增殖化细胞系。获自无限增殖化细胞系的细胞培养物的特定实例是人ptec-tert-1、ciptec、hk-2、nki-2或人a549细胞系。表达egfr但不具有egf消耗的无限增殖化细胞系的实例是caco2细胞。在本发明的一个实施方案中,表达egfr的细胞系不是caco2细胞系或arpe19细胞系。给予药物可以通过本领域已知的常用方法,如被动扩散、电穿孔、超声波处理、细胞挤压、静水压力、纳米颗粒(例如,脂质体)、磁转染和细胞渗透肽,将待在本发明的方法中评价的药物给予细胞培养物。通常使用转染或通过称为剥裸(gymnosis)(也称为裸露递送,参见stein等,nar201038(1)e3或soifer等,methodsmolbiol2012,815:333-46)的方法来给予核酸分子,尽管也可以应用其他给予选择。在所述实例中,可以看到使用egf生物标志物预测体外肾毒性与是否通过转染或通过剥裸将反义寡核苷酸给予细胞培养物无关。给予的方式需要适用于细胞培养物的性质。一些细胞类型更易于通过剥裸吸收寡核苷酸,如针对ptec类型细胞系证明的,而其他细胞系更适用于转染,如针对a549细胞系看到的。因此将给予方式进行优化,使得针对特定的细胞培养物研究最佳的给予方式。在本发明的一个实施方案中,通过剥裸给予核酸分子,特别是如果核酸分子是单链寡核苷酸,如反义寡核苷酸,或单链rnai剂。从5年多前发现开始,剥裸递送已经成为了寡核苷酸研究中使用的标准工具,并且是本领域充分确定的术语。通常,寡核苷酸的剥裸递送利用约1μm至约1000μm的寡核苷酸浓度,如约5μm至约100μm,如约10μm至约50μm,如约20μm至约40μm,如约25-35μm。合适地,可以将寡核苷酸给予例如在pbs中的细胞培养物,以获得1-100μm的终浓度,如5-50μm,如10或30μm。剥裸递送的优势之一是其类似于在体内发生的递送,因为不需要另外的化学物质来将寡核苷酸运入细胞中。在本发明的另一个实施方案中,通过转染给予核酸分子,这种方法对于单链和双链寡核苷酸都适用。可以使用本领域已知的标准方法来进行转染,如脂转染、阳离子聚合物、磷酸钙等。通常,转染利用0.5ng至50ng的寡核苷酸浓度,如1ng至25ng,如2ng至15ng,如约3ng至10ng。合适地,可以将寡核苷酸给予例如pbs中的细胞培养物,以获得对应于上述那些的终浓度。孵育时间药物给予和相关生物标志物测量之间的时间是孵育时间或培养时间。对于尽可能有效的体外毒性测定法,读出需要是可重复的并且一致地预测具有潜在毒性问题的化合物。另一个相关参数是进行测定法花费的时间,越快进行,通量越高,如果本发明的方法用于筛选大的药物文库,这是高度相关的。根据细胞培养物和给予方法,将孵育时间接受优化。在本发明的一个实施方案中,使用药物的孵育时间为1至9天,如2至8天,3至7天,或4至6天,特别是2至6天,或3至6天,如2、3、4、5、6、7、8或9天。发明人已经发现了对于胞外生物标志物egf和kim-1,使用2至6天孵育时间可以获得足够的可重复性,特别是在给予后第3天和第6天的生物标志物测量显示出良好的结果。在本发明的一个实施方案中,使用剥裸将浓度为50至100μm的单链寡核苷酸给予细胞培养物并且孵育3至6天,且在这些时间测量egf和/或kim-1。在其中测量胞内生物标志物atp的情况中,使用药物的孵育时间为5至10天,如6至9天,如6、7、8或9天。在其中使用转染进行给予的实施方案中,使用核酸分子的孵育时间为1至4天,如2至3天,如1、2、3或4天。在本发明的一个实施方案中,使用转染将1至20ng浓度的核酸分子给予细胞培养物,并且孵育1至3天,并且在这些时间测量egf和/或kim-1。哺乳动物细胞的细胞培养通常在或约37℃进行,并且可以进一步包括外源性co2,由此保持在5%co2或约5%co2的环境中。培养物中的细胞通常需要有规律的新鲜培养基,如每2至4天。如果孵育时间超过3至4天,培养基可以每3至4天更换。新鲜培养基优选含有对应于第0天给予浓度的药物浓度,以确保孵育时间过程中药物的持续存在。本发明的优点之一是预测性毒性标志物的早期读出,在孵育阶段开始后相对短时间地读出,并且使用的生物标志物提供了可靠的信号。筛选变体文库来鉴定具有预测的体内降低的毒性特征的子药物:本发明提供了从药物文库筛选一种或多种适用于体内施与哺乳动物的药物的方法,所述方法包括步骤:a)获得药物文库b)将药物文库的每个成员给予表达表皮生长因子受体(egfr)的细胞培养物,并且其中培养基含有至少4ng/ml的表皮生长因子(egf);c)并且在体外培养细胞一段时间;d)测量至少一种肾毒性生物标志物的含量;和e)选择其中毒性等级低于6的一种或多种药物。在本发明的一个实施方案中,与毒性参照物质或亲本药物相比时,步骤e)中选定的药物的治疗指数降低。选定的药物可以任选体内施与哺乳动物,以证实化合物没有引发肾毒性。特别地,大鼠对肾毒性核酸分子是高度敏感的,并且因此是用于证实使用本发明的方法获得的预测的相关物种。自然地,本文中提及的其他哺乳动物物种也可以是相关的。在一些实施方案中,药物文库是由rnai剂、反义寡核苷酸或适体组成的核酸分子文库。在一个实施方案中,核酸分子文库具有不同的核碱基序列,例如,它们可以是跨越靶序列(例如,mrna)设计的核酸分子文库,例如,通过mrna基因行走产生的反义寡核苷酸或rnai剂的文库。在一个实施方案中,核酸分子文库的成员具有在连续的序列段中具有相同的核碱基序列,其中连续的序列段短于核酸分子,文库中具有至少一个分子子集。这样的“热点”文库可以例如是跨越例如50至100个核碱基长度的靶序列设计的核酸分子的文库(例如,靶rna上的热点),其中文库的成员将沿着这个序列有至少4至14个核碱基,如5至12个核碱基,如6至10个核碱基的重叠。文库例如可以是鉴定具有靶向鉴定的热点的最高ti的寡核苷酸的反义寡核苷酸或rnai剂的文库。在一些实施方案中,药物文库是亲本核酸分子的核酸分子变体(子核酸分子)的文库,其中亲本核酸分子是有毒的,如肾毒性的,并且其中步骤d)鉴定一个或多个毒性低于亲本核酸分子的核酸分子变体;其中核酸分子变体保留亲本核酸分子(如反义寡核苷酸)的核心核碱基序列。在一些实施方案中,子核酸分子可以与亲本核酸分子是相同长度的,并且保留相同的核碱基序列。然而,设想的是,在一些实施方案中,子核酸分子可以是截短的,如通过去除5’和/或3’末端核苷酸,或在一些实施方案中,可以在5’和/或3’端具有另外的核苷酸。去除一个或多个末端高亲和性核苷,如lna核苷,允许维持核酸分子对rna靶的亲和性,同样将一个或多个lna核苷插入缺口区中将提高对rna靶的亲和性。设想的是,在一些实施方案中,子核酸分子文库可以包含具有不同侧翼区的变体,一些是截短的,一些具有另外的核苷,一些具有(如相对rna靶测量的)移动了一个或两个核苷的序列,一些在侧翼中具有另外的高亲和性核苷,因此文库是具有异源硫代磷酸酯核苷间键的立体限定的硫代磷酸酯寡核苷酸的复杂文库,由此允许同时选择与亲本核酸分子相比具有降低毒性的子核酸分子。亲本和子核酸分子共享共有的核心核碱基序列。共有的核心核碱基序列(或连续的核碱基序列)通常为至少10个核碱基长,如至少11个,至少12个,至少13个,至少14个,至少15个,或至少16个核碱基长,并且在一些实施方案中,可以是亲本核酸分子的相同核碱基序列。在一些实施方案中,亲本和(至少一部分)子核酸分子在跨越核酸分子长度中具有相同的核碱基序列。然而设想了在一些实施方案中,一部分子核酸分子可以包含另外的5’或3’核苷,如另外的1、2或3个5’或3’核苷。额外地或备选地,在一些实施方案中,一部分子核酸分子相对于亲本是截短的,例如,可以在5’或3’端包含1、2或3nt截短。在一些实施方案中,(一部分)子核酸分子核碱基序列的另外的核碱基或截短是单个核碱基添加或截短。在一些实施方案中,子寡核苷酸,或其一部分,与靶序列比对时,与亲本核酸相比,移动单个核碱基,或2或3个核碱基(实际上一端截短,而在另一端添加)。其他核苷酸保留与靶核酸序列的互补性。在一些实施方案中,核酸分子变体(子核酸分子)的文库的一个或多个设计参数不同于亲本核酸分子。设计参数可以选自i)一个或多个立体限定的硫代硫酸酯核苷间键的存在;ii)缺口大小的变化;iii)引入缺口打破者;vi)翼大小的改变;v)引入2’糖修饰的核苷;和vi)在翼中引入至少两个不同的2’修饰核苷的翼中的2’糖修饰的核苷组成的变化,如lna核苷和2’取代的核苷,特别是lna和moe。在一些实施方案中,核酸分子变体是寡核苷酸变体。在一些实施方案中,寡核苷酸变体是反义寡核苷酸。在一些实施方案中,反义寡核苷酸是缺口聚物寡核苷酸。在一些实施方案中,寡核苷酸变体是rnai剂。在一些实施方案中,寡核苷酸变体与亲本寡核苷酸的差异在于存在一个或多个立体限定的硫代磷酸酯核苷间键。在一些实施方案中,寡核苷酸变体是lna寡核苷酸。在一些实施方案中,子反义寡核苷酸的文库是或包含具有不同缺口聚物设计的子寡核苷酸群,任选包括不同的混合翼缺口聚物设计、缺口打破者设计、缺口长度和侧翼长度。本发明的方法可以用于鉴定具有降低的体内毒性(如肾毒性)的立体限定的核酸分子,特别是立体限定的反义寡核苷酸。本发明因此提供了使用立体限定的硫代磷酸酯核苷间键降低反义寡核苷酸(亲本寡核苷酸)的毒性的特定方法,其包括步骤:a)产生立体限定的寡核苷酸变体(子寡核苷酸)的文库,保留亲本寡核苷酸的核心核碱基序列;b)在表达表皮生长因子受体(egfr)的细胞培养物中筛选步骤a)中产生的文库并且其中培养基含有至少4ng/ml表皮生长因子(egf)(按照本文中所述的本发明的方法);和c)鉴定文库中存在的与亲本寡核苷酸相比在细胞培养物中具有降低毒性的一个或多个立体限定的变体。任选地,重复所述方法(反复筛选),例如,使得将通过所述方法鉴定的一个或多个立体限定的变体用作下一轮筛选方法中的亲本寡核苷酸。在本发明的方法中,步骤b)中产生的文库的每个成员包含至少一个不同于亲本的立体限定的硫代磷酸酯核苷间键。本发明的方法可以进一步包括另外的制造一个或多个使用本发明的方法之一选定的具有降低毒性的核酸分子变体的随后步骤。在一些实施方案中,随后的制造是超过1g的规模,如超过10g。在一些实施方案中,以低于1g,如低于0.5g,如低于0.1g的规模进行用于体内或体外筛选步骤的寡核苷酸的合成。在一些实施方案中,所述方法进一步包括测定药物文库或步骤c)或e)中鉴定的文库中的一个或多个选定的药物的体外或体内功效的步骤。本发明提供了用于预测反义寡核苷酸(如lna寡核苷酸)的体内肾毒性(例如,可能性)的方法,所述方法包括将寡核苷酸给予表达表皮生长因子受体(egfr)的细胞培养物的步骤,且其中培养基包含至少4ng/ml表皮生长因子(egf),在寡核苷酸的存在下孵育细胞,例如,孵育2-9天的时间段,如2-6天,如3天,并且随后测量至少一种生物标志物的毒性,如本文中所述的那些,例如,通过测量释放至培养基中的egf和/或kim-1的含量,并且任选测量胞内atp水平。合适地,细胞atp水平的降低表示肾毒性寡核苷酸,并且培养基中的egf或kim-1的升高表示肾毒性寡核苷酸。本发明提供了体外测定法测定药物的肾毒性(例如,可能性)的用途,所述药物如核酸分子,如反义寡核苷酸,如lna寡核苷酸。将认识到,在一些实施方案中,用于预测(或测定)体内毒性(例如,肾毒性)的方法可以用于鉴定亲本寡核苷酸的立体限定变体,其中立体限定变体具有降低的体外或体内毒性。补充毒性测定法除了本申请中所述的用于预测药物的体内肾毒性的方法,所述方法可以进一步结合用于预测与药物(特别是治疗性寡核苷酸,如反义寡核苷酸)研发相关的其他毒性的方法。在一些实施方案中,本申请中公开或要求的用于预测肾毒性的方法可以结合用于预测药物(特别是寡核苷酸)的免疫毒性的方法。一种用于预测药物(特别是寡核苷酸)的免疫毒性的方法包括测量接受药物的血液样本中的至少一种补充生物标志物和/或至少一种(如至少两种)细胞因子生物标志物。在特别的实施方案中,通过测量接受药物(特别是寡核苷酸)的血液样本中的至少一种补充生物标志物和至少两种细胞因子生物标志物来预测免疫毒性。在一些实施方案中,本发明的方法进一步包括或提供预测免疫毒性的方法,包括步骤a)将药物(特别是寡核苷酸)给予人血液(分离自身体);b)将样品孵育30分钟至8小时;c)停止反应;和d)测量至少两种、三种、四种、五种或全部以下的生物标志物:i)补体生物标志物c3a和c5a,和/或ii)细胞因子生物标志物白细胞介素6(il6)、白细胞介素8(il8)、肿瘤坏死因子α(tnfα)和单核细胞化学引诱物蛋白-1(mcp1);其中至少两种生物标志物与对照相比高于约2倍的平均提高表示药物(如寡核苷酸)的体内免疫毒性。步骤a)的血液样本通常获自至少一名健康的人受试者,如至少两名或至少三名健康的人受试者。通常,血液样本没有混合。在一些实施方案中,结合至少两种选自白细胞介素6(il6)、白细胞介素8(il8)、肿瘤坏死因子α(tnfα)和单核细胞化学引诱物蛋白-1(mcp1)的细胞因子生物标志物来测量至少一种补体生物标志物c3a和c5a。在一些实施方案中,至少测量了细胞因子生物标志物白细胞介素8(il8)和单核细胞化学引诱物蛋白-1(mcp1)。在一些实施方案中,使用了来自至少3名供体的血液样本。通常,血液获自健康供体(即,从身体分离)。在一些实施方案中,将来自3个不同血液样本的测量平均,以获得免疫毒性读出。在一些实施方案中,本申请中公开或要求的方法可以结合用于预测药物(特别是寡核苷酸)肝毒性的方法。一种用于预测寡核苷酸的体内肝毒性的方法描述于wo2017/067970中,在此并入本文作为参考。简而言之,一种预测寡核苷酸的体内肝毒性的方法包括步骤a)将寡核苷酸在体外给予细胞培养基中的原代哺乳动物肝细胞群(或源自诱导的多潜能干细胞的肝细胞群);b)将细胞在细胞培养基中体外培养一段时间,如1至14天,特别是2至7天;和c)随后测量释放至培养基中的乳酸脱氢酶(ldh)的水平,和/或测量细胞atp水平;其中与对照比较细胞培养基中的乳酸脱氢酶的提高,如20%提高,或与对照相比细胞atp水平的降低,如20%降低,表示寡核苷酸是或预测是在哺乳动物中体内肝毒性的。可以补充肝毒性预测的更多生物标志物是释放至培养基中的微rna-122,其中细胞培养基中的微rna-122的提高预测了肝毒性,以及胞内谷胱甘肽(gsh)水平,其中ghs水平的降低预测了肝毒性。在本发明的一些实施方案中,将用于预测体内肾毒性的方法结合预测体内免疫毒性的方法和预测体内肝毒性的方法,如以上所述的方法。药物组合物在再一个方面中,本发明提供了通过本发明的方法获得的药物或其盐。在再一个方面中,通过本发明的方法获得的药物或其盐可以配制成药物组合物,其包含上述任一种药物和可药用稀释剂、运载体、盐和/或佐剂。可药用稀释剂包括磷酸盐缓冲盐水(pbs),并且可药用盐包括但不限于钠盐和钾盐。在一些实施方案中,可药用稀释剂是无菌磷酸盐缓冲盐水。在一些实施方案中,将寡核苷酸以50-300μm溶液的浓度用于可药用稀释剂中。用于本发明中的合适制剂可以在remington'spharmaceuticalsciences,mackpublishingcompany,philadelphia,pa.,第17版,1985中找到。对于用于药物递送的简短综述,参见,例如,langer(science249:1527-1533,1990)。wo2007/03109提供了更多合适的和优选的可药用稀释剂、运载体和佐剂的实例(在此并入本文作为参考)。wo2007/031091还提供了合适的剂量、配方、给药途径、组合物、剂型、与其他治疗剂的组合、前药制剂。通过本发明的方法获得的药物可以与可药用活性或惰性物质混合,用于制备药物组合物或制剂。组合物和用于配制药物组合物的方法取决于许多标准,包括但不限于给药途径、疾病程度或待给药的剂量。可以通过常规灭菌技术将这些组合物灭菌,或可以是无菌过滤的。将所得到的水溶液包装,按现状使用,或冷冻干燥,在给药前,将冻干的制备物与无菌含水溶媒混合。制备物的ph通常为3至11,更优选5至9,或6至8,并且最优选7至8,如7至7.5。将所得到的固体形式的组合物以多个单剂量单位包装,每个含有固定量的上述一种或多种药剂,如在片剂或胶囊的密封包装中。固体形式的组合物还可以包装在用于灵活定量的容器中,如针对局部施用的霜剂或膏剂设计的可挤压管。本发明提供了用于治疗或预防疾病的方法,包括将治疗或预防有效量的通过本发明的方法获得的药物或其盐或本发明的药物组合物给药于患有或易感疾病的受试者。本发明还涉及通过本发明的方法获得的药物或其盐或如本文中定义的药物组合物用作药物。通过本发明的方法获得的药物或其盐或根据本发明的药物组合物通常以有效量来给药。本发明还提供了通过本发明的方法获得的药物或其盐的用途,用于制造治疗本文中提及的疾病的药物,或用于本文中提及的疾病的治疗方法。发明的实施方案本发明的以下实施方案结合本文中所述的任何其他实施方案来使用。1.用于预测药物在哺乳动物中的体内肾毒性的体外方法,所述方法包括步骤:a.在含有至少4ng/ml表皮生长因子(egf)的合适的细胞培养基中培养表达表皮生长因子受体(egfr)的细胞;b.将药物给予所述细胞培养物;c.孵育细胞一段时间;和d.随后测量上清液中的egf水平;其中上清液中egf的提高表示药物是或预测是与肾毒性相关。2.根据实施方案1的方法,其中将上清液中的egf水平与获自用溶媒对照或无毒参照药物处理的细胞的参照值相比,其中所述无毒药物已经证实为在体内是无毒的。3.根据实施方案2的方法,其中无毒参照药物是由cgtcagtatgcgaatc(seqidno:1)组成的寡核苷酸化合物,其中小写字母表示dna单元,粗体大写字母表示β-d-氧基-lna单元,所有lnac是5’甲基c以及所有核苷间键是硫代磷酸酯键。4.根据实施方案3的方法,其中上清液中的egf水平相对于溶媒对照或无毒参照值高于200%是药物肾毒性的预测。5.根据实施方案2至4的方法,其中将上清液中的egf水平进一步与获自用肾毒性参照药物处理的细胞的第二参照值相比,其中所述肾毒性参照药物已经证实了引起体内肾毒性。6.根据实施方案5的方法,其中作为无毒参照药物或溶媒对照和肾毒性参照药物之间的差异来确定测定窗口(aw)。7.根据实施方案5的方法,其中所述毒性参照药物是由gctgtgtgagcttgg(seqidno:4)组成的寡核苷酸化合物,其中小写字母表示dna单元,粗体大写字母表示β-d-氧基-lna单元,所有lnac是5’甲基c以及所有核苷间键是硫代磷酸酯键。8.根据实施方案5至7的方法,其中根据下式确定毒性等级9.根据实施方案2至8中任一个所述的方法,其中毒性等级高于6是药物肾毒性的预测,如高于20,如高于50。10.根据实施方案1至9中任一项所述的方法,其中步骤d)进一步包括测量胞内三磷酸腺苷(atp)水平;其中胞内atp水平的降低表示药物是或预测是与肾毒性相关。11.根据实施方案10的方法,其中胞内atp的水平相对于盐水或无毒参照值低于80%是药物肾毒性的预测。12.根据实施方案1至11中任一个所述的方法,其中步骤d)进一步包括测量胞外肾损伤分子-1(kim-1)蛋白或胞内mrna水平,其中kim-1水平的提高表示药物是或预测是与肾毒性相关。13.根据实施方案12的方法,其中相对于盐水或无毒参照值高于200%的水平是药物肾毒性的预测。14.根据实施方案1至13中任一个所述的方法,其中表达egfr的细胞在72小时内消耗培养基至少50%的egf。15.根据实施方案1至14任一个的方法,其中表达egfr的细胞选自上皮细胞、内皮细胞、间充质细胞、神经外胚层细胞和肝细胞。16.根据实施方案1至15任一个的方法,其中表达egfr的细胞是上皮细胞培养物或肝细胞培养物。17.根据实施方案16的方法,其中细胞是选自啮齿动物原代细胞(如小鼠或大鼠原代细胞);猪(例如,迷你猪)细胞和灵长类原代细胞,如猴(例如,食蟹猴)、或人原代细胞的原代细胞培养物的形式。18.根据实施方案17的方法,其中原代细胞培养物获自大鼠或人上皮细胞或肝细胞。19.根据实施方案16至18任一个的方法,其中上皮细胞培养物是肾上皮细胞培养物或肺上皮细胞培养物。20.根据实施方案19的方法,其中肾上皮细胞选自近端小管上皮细胞、远端小管上皮细胞和集合管上皮细胞。21.根据实施方案19或20中任一项所述的方法,其中细胞培养物从人ptec或大鼠ptec细胞制得。22.根据实施方案1至16或19或20中任一个所述的方法,其中表达egfr的细胞培养自无限增殖化细胞系。23.根据实施方案22的方法,其中细胞系不是caco2或arpe19。24.根据实施方案22的方法,其中细胞培养物获自人ptec-tert-1、ciptec、hk-2、nki-2或人a549细胞系。25.根据实施方案1至24中任一个所述的方法,其中细胞培养基含有5至15ng/ml,如8至15ng/ml表皮生长因子(egf),如约10ng/ml。26.根据实施方案1至24中任一个所述的方法,其中用药物孵育的时间段为2至6天,如大约3天。27.根据实施方案1至26中任一个所述的方法,其中药物选自基于核酸的分子、化疗剂;氨基糖苷类;抗细菌剂、抗病毒剂;抗真菌剂、抗炎剂和免疫抑制剂。28.根据实施方案1至27中任一个所述的方法,其中药物是选自rnai剂、反义寡核苷酸或适体的核酸分子。29.根据实施方案1至28中任一个所述的方法,其中药物是反义寡核苷酸。30.根据实施方案29的方法,其中在转染剂不存在下将寡核苷酸给予细胞培养物,即通过称为剥裸(gymnosis)的方法给予。31.根据实施方案28的方法,其中在转染剂存在下将寡核苷酸给予细胞培养物。32.根据实施方案28至31中任一个所述的方法,其中核酸分子包含一个或多个2’糖修饰的核苷。33.根据实施方案32的方法,其中一个或多个2’糖修饰的核苷独立地选自2’-o-烷基-rna、2’-o-甲基-rna、2’-烷氧基-rna、2’-o-甲氧基乙基-rna、2’-氨基-dna、2’-氟-dna、阿糖核酸(ana)、2’-氟-ana和lna核苷。34.根据实施方案32或33的方法,其中一个或多个2’糖修饰的核苷是lna核苷。35.根据实施方案33或34的方法,其中lna核苷选自β-d-氧基-lna、α-l-氧基-lna、β-d-氨基-lna、α-l-氨基-lna、β-d-硫代-lna、α-l-硫代-lna、(s)cet、(r)cet、β-d-ena或α-l-ena。36.根据实施方案28至35中任一项所述的方法,其中核酸分子包含至少一个修饰的核苷间键。37.根据实施方案36的方法,其中连续核苷酸序列内的核苷间键是硫代磷酸酯核苷间键。38.根据实施方案28至37中任一个所述的方法,其中反义寡核苷酸能够募集rnaseh。39.根据实施方案38的方法,其中反义寡核苷酸是缺口聚物。40.根据实施方案38或39的方法,其中寡核苷酸是式5’-f-g-f’-3’的缺口聚物,其中区域f和f’独立地包含1-7个修饰的核苷并且g是能够募集rnaseh的6至16个核苷的区域。41.根据实施方案12或13的方法,其中kim-1的提高是根据实施方案28至40任一个的核酸分子的肾毒性的预测,即使egf水平没有提高。42.根据实施方案1至40中任一个所述的方法,其中体内肾毒性是急性肾损伤和/或小管退化。43.一种从药物文库选择用于体内施与哺乳动物的一种或多种药物的方法,所述方法包括步骤:a.获得药物文库;b.将药物文库的每个成员给予表达表皮生长因子受体(egfr)的细胞培养物并且其中培养基含有至少4ng/ml表皮生长因子(egf),如按照实施方案14至25中的任一个;c.在体外培养细胞一段时间,如按照实施方案26;d.针对每种药物测量胞内egf的含量,如按照实施方案1至13中的任一个;和e.选择一种或多种药物,其中根据实施方案8测定时,毒性等级低于6。44.根据实施方案43的方法,其中与毒性参照物质或亲本药物相比时,选定的药物的治疗指数降低。45.根据实施方案43或44的方法,其中药物文库是按照实施方案28至40中任一个所述的核酸分子的文库。46.根据实施方案44的方法,其中核酸分子文库是亲本核酸分子的核酸分子变体(子核酸分子)的文库,其中亲本核酸分子是有毒的,如肾毒性的,并且其中步骤d)鉴定了毒性低于亲本核酸分子的一个或多个核酸分子变体;其中核酸分子变体保留亲本核酸分子的核碱基序列。47.根据实施方案44至46中任一个所述的方法,其中亲本核酸分子是按照实施方案28至40中任一个所述的核酸分子,特别是反义寡核苷酸。48.根据实施方案44至47中任一个所述的方法,其中核酸分子变体的文库包含通过设计不同于亲本核酸分子的子核酸分子群。49.根据实施方案44至48的方法,其中核酸分子变体与亲本核酸分子的一个或多个设计参数不同,所述设计参数选自i.一个或多个立体限定的硫代磷酸酯核苷间键的存在;ii.缺口大小的改变;iii.缺口打破者的引入;iv.翼大小的改变;v.翼中2’糖修饰的核苷的改变;和vi.在翼中具有至少两个不同的2’修饰的核苷的混合翼缺口聚物,如lna核苷和2’取代的核苷,特别是moe。50.根据实施方案44至49的方法,其中选择一种或多种用于体内施与哺乳动物的药物进一步基于来自体外免疫毒性测定法和/或体外肝毒性测定法的结果,所述体外测定法适用于预测体内免疫毒性和/或体内肝毒性。51.通过根据实施方案43至50中任一项所述的方法获得的药物。52.包含实施方案51的药物和可药用稀释剂、溶剂、运载体、盐和/或佐剂的药物组合物。53.实施方案51的药物或实施方案52的药物组合物,用于药物中。实施例方法和材料化合物表1实施例中使用的寡核苷酸的列表对于化合物,小写字母表示dna单元,大写字母表示β-d-氧基-lna单元。所有lnac单元是5’甲基c并且所有核苷间键是硫代磷酸酯键。rp立体限定的硫代磷酸酯键通过下标r来表示。sp立体限定的硫代磷酸酯键通过下标s来表示。粗斜体的小写字母表示moe单元。在化合物20-1中,所有c单元(dna和moe)是5’甲基c。seqidno是指化合物的核碱基序列。测量体内肾毒性将获自charlesriver实验室的7至8周大的有意繁育的(purposebred)wistarhancrl:wi(han)雄性大鼠基于体重分成4组(表1,实验a)或8组(表1,实验b),并且在给药前适应环境至少5天。将动物圈养在标准环境条件下(22±2℃,相对湿度50±20%,光/暗周期12h/12h,球状食物和水随意使用),并且在aaalac公认的设施中提供富足的环境,并且定期和小心地监控。所有程序根据依照地方性法规并且依照机构动物管理和使用委员会(institutionalanimalcareandusecommittee)授予的动物许可。在等渗无菌盐水中配制测试化合物,无菌过滤(0.22μm),并且在第1天和第8天在肩胛间区以40mg/kg给药(2.5ml/kg)。对照组动物接受了盐水作为溶媒对照。在第15天,动物口服自来水(10ml/kg),并且在代谢笼中在冰上收集尿液6小时。测量了尿蛋白水平(autionmaxax-4280)和尿肾损伤生物标志物(multiplexmapratkidneytoxicitymagneticbeadpanel2)。在第15天,通过腹膜内注射戊巴比妥并且放血,将大鼠牺牲。收集肾皮质样品,并且通过浸没在10%中性缓冲的福尔马林中固定,包埋在石蜡中,切片成5μm,并且用苏木精和伊红(h&)染色。随后使用aperioscanscopeat(leicabiosystems)扫描仪在20×放大倍数下,将h&e切片扫描,并且随后通过imagescope软件从扫描的图像捕获图片。评价以下的一个或多个参数:肾脏重量、血清肌酸酐、尿蛋白、尿中的kim-1蛋白、kim-1mrna、肾退化/再生和相对的肾小管毒性等级。细胞培养物人原代近端小管上皮细胞(ptec)细胞培养物根据制造商的说明,在ptec培养基[dmem/f12,不含酚红(thermofisherscientific11039021),含有1%青霉素-链霉素溶液(thermofisherscientific15140122)、10mmhepes(thermofisherscientific15630056)、5ug/ml胰岛素和5ug/ml转铁蛋白和8.65ng/ml亚硒酸钠(全部来自100×浓缩储液,thermofisherscientific41400045)、100nm氢化可的松(sigmah6909),3.5ug/ml抗坏血酸(sigmaa4403)、25ng/ml前列腺素e1(sigmap5516)、3.25pg/ml三碘代-l-甲状腺原氨酸(sigmat6397)、10ng/ml人重组表皮生长因子(egf,r&dsystems236-eg-200)、100μg/ml遗传霉素(g418硫酸盐,thermofisherscientific10131027)]中培养原代ptec(sciencecellresearchlabotories,目录号#4100)。在用寡核苷酸处理之前,将原代ptec细胞分别以40000和20000个细胞/孔的密度接种于胶原蛋白i包被的96孔平板(corning,356407)中的ptec培养基中,并生长直至汇合。人ptec-tert1细胞培养物根据制造商的说明,在ptec培养基[dmem/f12,不含酚红(thermofisherscientific11039021),含有1%青霉素-链霉素溶液(thermofisherscientific15140122)、10mmhepes(thermofisherscientific15630056)、5ug/ml胰岛素和5ug/ml转铁蛋白和8.65ng/ml亚硒酸钠(全部来自100×浓缩储液,thermofisherscientific41400045)、100nm氢化可的松(sigmah6909),3.5ug/ml抗坏血酸(sigmaa4403)、25ng/ml前列腺素e1(sigmap5516)、3.25pg/ml三碘代-l-甲状腺原氨酸(sigmat6397)、10ng/ml人重组表皮生长因子(egf,r&dsystems236-eg-200)、100μg/ml遗传霉素(g418硫酸盐,thermofisherscientific10131027)]中培养ptec-tert1(evercytegmbh,奥地利)。在用寡核苷酸处理之前,将ptec-tert1分别以40000和20000个细胞/孔的密度接种于胶原蛋白i包被的96孔平板(corning,356407)中的ptec培养基中,并生长直至汇合。大鼠原代近端小管上皮细胞(ptec)细胞培养物通过bruce等,methodsmolbiol1001,53-64(2013)描述的简化程序,来制备近端小管上皮细胞。从一只麻醉的雄性wistar大鼠(年龄:10-18周)取出肾脏并且在冷的1×浓缩hanks平衡盐溶液(hbss,thermofisherscientific14065049)中洗涤。将器官去除被膜,并将无髓质的肾皮质外纹(outerstripe)切成1至2mm3小块。在25ml含有2mg/ml胶原酶a(roche10103586001)和10ug/mldnasei(roche10104159001)的hbss中,将组织小块在37℃,连续温和振荡消化60分钟。通过添加25ml含有2%牛血清白蛋白(bsa,fractionv,roche10735086001)的冰冷hbss来停止反应。将解离的组织首先通过200μm细胞滤网(pluriselect43-50200)并且随后通过100um细胞滤网(pluriselect43-50100)来连续过滤;将含有单细胞和小管碎片的滤过物洗涤并在不含mgcl2和cacl2的hbss溶液(hbss-/-,thermofisherscientific14175053)中离心三次(100g)。将沉淀物重悬浮于25mlhbss-/-中,并且与25ml30%的optiprep溶液(5体积的60%optiprep密度梯度培养基(sigmad1556)、4体积的无菌h2o和1体积的不含mgcl2和cacl2的10×浓缩磷酸盐缓冲盐水(pbs-/-,thermofisherscientific14200-067)的混合物)混合,以获得细胞和小管碎片在15%optiprep中的悬浮液。将悬浮液离心(800g,室温20分钟)。收集试管上三分之一的细胞带;将试管底物的沉淀物中的细胞丢弃。将收获的细胞在大鼠ptec培养基[1体积dmem(thermofisherscientific11966025)、1体积ham’sf-12营养混合物(thermofisherscientific21765029)、2%胎牛血清(fbs,thermofisherscientific16000044)、1%青霉素-链霉素溶液(thermofisherscientific15140122)、10mmhepes(thermofisherscientific15630056)、5ug/ml胰岛素和5ug/ml转铁蛋白和8.65ng/ml亚硒酸钠(全部来自100×浓缩储液,thermofisherscientific41400045)、100nm氢化可的松(sigmah6909),3.5ug/ml抗坏血酸(sigmaa4403)、25ng/ml前列腺素e1(sigmap5516)、3.25pg/ml三碘代-l-甲状腺原氨酸(sigmat6397)和10ng/ml大鼠重组表皮生长因子(egf,r&dsystems3214-eg-100/cf)]中洗涤并离心三次(400g,室温下5min)。最终离心步骤后,将细胞制备物重悬浮于50ml大鼠ptec培养基中,并分配至三个胶原蛋白i包被的150cm2细胞培养瓶中(corning354486),接着孵育4天,头2天后更换培养基。在用寡核苷酸处理之前,通过用胰蛋白酶-edta溶液(thermofisherscientific25200056)分离,从培养瓶收集扩增的大鼠ptec细胞,在大鼠ptec培养基中洗涤,并以36000个细胞/孔的密度接种于胶原蛋白i包被的96孔平板中(corning,356407)的大鼠ptec培养基中,生长直至汇合。原代人肝细胞将冷冻保存的人肝细胞以大约1×106个细胞/ml的密度悬浮于william氏培养基e(wme)中(不含酚红)(sigmaw-1878),补充了10%胎牛血清、青霉素(100u/ml)、链霉素(0.1mg/ml)),并以0.4×105个细胞/孔的密度接种于胶原蛋白覆盖的96孔平板(bectondickinsonag,allschwil,瑞士)中。用寡核苷酸处理开始前,将细胞预培养3至4h,允许贴壁至细胞培养板。在用寡核苷酸处理之前,接种培养基替换为90μl无血清wme。a549细胞(腺癌人肺泡基底上皮细胞)根据制造商的说明在a549培养基[f-12knut混合物(gibco,21127-022),1%青霉素/链霉素(gibco:15140-122),10%fbs(gibco16000-044)]中培养a549细胞(atccccl-185)。在用寡核苷酸处理之前,将a549细胞以3000个细胞/孔接种于a549常规培养基中并生长直至60%汇合。对于脂转染,根据制造商的说明,使用脂质转染胺(invitrogen,11668-019),用1或10ng寡核苷酸转染细胞。24小时后,将培养基换成含有10ng/mlegf和2%fcs的a549常规培养基。72小时后,收获细胞并测量atp,并且将上清液储存在-20℃,用于egf分析。使用剥裸的寡核苷酸处理将寡核苷酸溶解于pbs中并以给定的浓度(如10、30或100μm)加入细胞培养物中。每个孔的总体积为100μl。将pbs用作溶媒对照。寡核苷酸处理后待分析胞内atp时,每3天更换培养基,包括添加第0天相同浓度的寡核苷酸。寡核苷酸处理后分析胞外标志物(如egf和kim-1)时,收集培养基并储存在-20℃直至进一步分析。收集第一次寡核苷酸处理生长超过3天的细胞的培养基并且每3天替换包括新鲜寡核苷酸的培养基。将收集的培养基储存在-20℃。atp测定法对于胞内atp水平的测定,根据制造商的说明,使用celltiter-发光细胞成活力测定法(g7571,promegacorporation,madisonwi,usa)。每个样品重复测试三次。除非另外指出,在寡核苷酸处理后第9天,测量了胞内atp水平。egf测定法对于人egf的分析,将细胞上清液在冰上融化,在样品稀释缓冲液(biorad目录号#m60-009rdpd)中1:2和1:10稀释并根据制造商的说明,使用人egf珠(bio-plexprotm人癌症生物标志物组2egfset#171bc603m)通过基于luminex的elisa分析,或接着使用bio-200systems(biorad)分析。将数据报告为三个孔的平均浓度和标准差。高于200%的egf水平的提高认为是肾毒性的预测。kim-1测定法对于人kim-1的分析,将细胞上清液在冰上融化,并在样品稀释缓冲液(biorad目录号#m60-009rdpd)中1:2和1:10稀释并根据制造商的说明,通过使用人kim-1珠(人肾损伤组4,milliporehki4mag-99k-kim1)的基于luminex的elisa分析,接着使用bio-200systems(biorad)发您西。数据报告为三个孔的平均浓度和标准差。高于200%的kim-1水平的提高认为是肾毒性的预测。实施例1寡核苷酸处理时细胞培养物的形态学变化这个实施例评价了寡核苷酸处理后对细胞培养物的形态学的影响。ptec-tert1细胞是如“材料和方法”部分中所述的培养物。为了模拟肾小管对循环的裸寡核苷酸的生理暴露(即,没有借助递送技术,在本文中称为剥裸或剥裸递送),将ptec-tert1细胞的汇合单层暴露于100μm寡核苷酸的水溶液。每三天替换培养基,在新鲜培养基中包括100μm新鲜寡核苷酸。在处理7天后,在亮场显微镜下研究处理过的细胞培养物中的形态学改变。图1显示了用寡核苷酸化合物1-1(在体内无害的)处理的ptec-tert1细胞与盐水处理的细胞相比没有显示出任何形态学变化。用寡核苷酸化合物3-1(中等体内毒性)处理的细胞形成不规则的圆顶和液泡,而用寡核苷酸化合物4-1(高体内毒性)处理的ptec-tert1细胞显示出扁平且稳定的外观。两种有毒的化合物都显示出形态学变化,然而较低毒性的化合物3-1产生比高毒性化合物4-1更明显的形态学变化。总之,有毒的寡核苷酸影响了形态学,然而使用形态学变化不能预测毒性的严重性。实施例2寡核苷酸对ptec和ptec-tert1细胞中的胞外egf的作用这个实施例评价了寡核苷酸暴露后对细胞培养基中的可溶性生物标志物egf的作用。根据“材料和方法”部分中描述的条件,培养了原代ptec细胞和无限增殖化的ptec-tert-1细胞系。在汇合时,按照“材料和方法”部分中所述的,用3、10、30或100μm浓度的寡核苷酸处理细胞。根据“材料和方法”部分中的egf测定法,测量了培养基中的egf浓度。结果显示于以下的表2中,并且表示三个相同处理的平均。表2.寡核苷酸处理后3或6天的egf浓度从这可以看出在第3天已经在原代ptec细胞中,有毒的寡核苷酸(化合物3-1和4-1)显示出在100μm寡核苷酸浓度下培养基中的可溶性egf与盐水以及无害化合物1-1相比时显著提高。在第6天,在原代ptec细胞中可以检测到降至10μm浓度的有毒化合物,而在ptec-tert1细胞中需要100μm。此外,对于第6天两种细胞系中的这些化合物,可以区分体内观察到的毒性的严重程度。ptec-tert1细胞由通过人端粒酶逆转录酶(tert)的催化亚基的稳定表达无限增殖化的人近端小管细胞组成。ptec-tert1研发为维持原代ptec细胞的形态和功能特性的细胞模型,并且另外具有优于由于端粒缩短具有有限寿命的原代细胞的复制优势。实施例3寡核苷酸对ptec-tert1细胞中的胞外egf的作用这个实施例评价了作为无限增殖化ptec-tert1细胞中的生物标志物egf是否可以用于区分体内观察的毒性等级,使用一种无害寡核苷酸和4种具有不同体内毒性等级的寡核苷酸。根据“材料和方法”部分中所述的条件,培养了无限增殖化的ptec-tert-1细胞系。汇合时,按照“材料和方法”部分中所述的,用3、10、30或100μm浓度的寡核苷酸处理细胞。根据“材料和方法”部分中所述的egf测定法,在处理后第3天、第7天和第10天,测量了egf浓度。结果显示于以下的表3中,并且表示三个相同处理的平均。表3.寡核苷酸处理后第3天、第7天和第10天的egf浓度从这可以看出在第3天,高度毒性的化合物(化合物4-1)显示出以33或100μm给药时,与其他化合物相比时,培养基中可溶性egf的显著提高。在第7天,可以检测培养基和降至33μm浓度的高度毒性的化合物,并且在100μm寡核苷酸下可以区分化合物之间的毒性严重程度。在第10天,更难以区分中等毒性寡核苷酸(化合物3-1)与低毒性寡核苷酸(化合物2-1)。然而,以100μm给药时,在第10天,两者都能与无害寡核苷酸(化合物1-1)区分,毒性更高的化合物4-1和5-1仍然能够充分区分。实施例4寡核苷酸对ptec-tert1细胞中的胞外egf的作用这个实施例评价了实施例3中的发现对于较大组的针对其他靶的寡核苷酸是否是可重现的。在相同研究中,还测量了两种其他生物标志物kim-1和胞内atp。根据“材料和方法”部分中所述的条件,培养了无限增殖化的ptec-tert-1细胞系。汇合时,按照“材料和方法”部分中所述的,用10、30或100μm浓度的寡核苷酸处理细胞。在第6天进行了egf和kim-1测量。对于atp测量,在寡核苷酸处理后第9天,收获了细胞。根据“材料和方法”部分中所述的测定法,测量了egf、kim-1和atp浓度。结果显示于以下的表4中,并且表示三个相同处理的平均。表4.第6天培养基中的egf和kim-1水平,寡核苷酸处理后第9天的胞内atp水平从表4可以看出egf生物标志物在100μm寡核苷酸剂量下能够预测出12种有毒化合物中的10种。kim-1只能预测出12种有毒化合物中的2种,然而,这些是egf生物标志物没有获取的化合物,表明kim-1可以补充用egf作为生物标志物获得的预测。atp作为生物标志物能够预测出12种有毒化合物中的5种。因此,atp生物标志物可以用于补充egf生物标志物,但没有像egf生物标志物那样地预测。结果也概括于图2中。实施例5寡核苷酸对原代肝细胞中的胞外egf的作用在以下实施例中,研究了表达功能性egfr的非肾细胞是否可以用于预测寡核苷酸的肾毒性。按照“材料和方法”部分中所述的培养了人原代肝细胞,培养基中添加了10ng/mlegf。汇合时,按照“材料和方法”部分中所述的,用10或100μm浓度的寡核苷酸处理细胞。在第3天获得了培养基,并且根据“材料和方法”部分中所述的测定法,测量了egf水平。结果显示于以下的表5中,并且表示三个相同处理的平均。表5.寡核苷酸处理后第3天,来自原代肝细胞的培养基中的egf水平从这可以看出,在100μm寡核苷酸下,使用原代肝细胞可以鉴定所有肾毒性化合物,并且严重毒性的化合物(4-1)可以与更中等毒性的化合物区分开来。实施例6寡核苷酸对转染细胞中的胞外egf的作用在这个实施例中,测试了比剥裸更适于用寡核苷酸转染的细胞系是否可以应用于预测肾毒性的方法中。按照“材料和方法”部分中所述的培养了a549细胞。50-60%汇合时,使用25μloptimem(gibco31985062)中的0.5μl脂质转染胺(invitrogen,11668-019)和相同体积optimem中的1ng或10ng寡核苷酸,将寡核苷酸转染至细胞中。将细胞在37℃孵育过夜。使用a459常规培养基+10ng/mlegf更换培养基。转染后48小时,收集培养基用于egf测量,并且收集细胞用于atp测量。结果显示于以下的表6中,并且表示三个相同处理的平均。表6.用寡核苷酸转染后48小时,来自a549细胞的egf和atp水平这些数据表明egf也可以用作转染的上皮细胞中的肾毒性的生物标志物。具有高毒性的寡核苷酸(4-1和8-1)在1ng已经是可检测的,而中等毒性的寡核苷酸(3-1)需要10ng。数据还表明atp在10ng寡核苷酸的剂量下可以用作转染的上皮细胞中的肾毒性的生物标志物。无害的寡核苷酸(2-1)显示出用10ng寡核苷酸转染时没有毒性的迹象,egf和atp作为生物标志物都是如此。潜在地,1至10ng的浓度,如5ng,可能更合适。实施例7:对用亲本寡核苷酸的设计变体处理的细胞中的胞外egf的作用在这个实施例中,筛选了与亲本化合物具有相同核苷酸序列但具有不同设计(在这种情况中,为给定位置的固定立体化学)的寡核苷酸变体的小文库,以选择具有毒性低于亲本化合物的寡核苷酸变体。已知亲本寡核苷酸在体内测试时引发中等毒性(化合物10-1)或高毒性(化合物8-1)。根据“材料和方法”部分中所述的条件,培养了无限增殖化的ptec-tert-1细胞系。汇合时,按照“材料和方法”部分中所述的,用10或30μm浓度的寡核苷酸处理细胞。根据“材料和方法”部分中所述的测定法,在处理后第6天测量了egf水平。结果显示于以下的表7中,并且表示三个相同处理的平均。根据下式计算了毒性等级:其中aw是以盐水参照和肾毒性参照化合物4-1之间的差异确定的测定窗口(即,aw=盐水-毒性参照)。表7.ptec-tert1细胞中6天后手性修饰的寡核苷酸文库的egf水平从这些数据,可以看出使用本测定法,可以筛选寡核苷酸变体文库并且鉴定与亲本分子相比时具有显著降低毒性等级的化合物。特别地,化合物10-2和10-4是用于进一步体内测试的合适候选物。实施例8ptec-tert-1细胞中的egf消耗这个实施例研究了ptec-tert1细胞中egf是怎样消耗的,并且还研究了培养基中的egf在寡核苷酸毒性的预测中的作用。将10ngegf/ml培养基加入细胞后72小时后来自未处理的ptec-tert1细胞培养基的egf消耗显示于表8中,并且清楚地显示了未处理的ptec-tert1细胞随着时间消耗了培养基中的egf。表8.ptec-tert1细胞中的egf消耗实施例9培养基中的egf的相关性按照“材料和方法”部分中所述的培养ptec-tert1细胞,并且在汇合时,在培养基中存在(10ng/ml)egf或不存在egf下,用25或100μm寡核苷酸处理细胞6天,来评价寡核苷酸的毒性时将egf加入培养基中的相关性。结果显示于表9中并且表示三个相同处理的平均。表9:评价寡核苷酸毒性时培养基中的egf的作用从这可以看出,培养基中不存在egf时,不可能看到用有毒寡核苷酸处理时对培养基中的egf水平的作用。另一方面,培养基中存在10ng/mlegf时,用有毒寡核苷酸处理的细胞中的egf水平在培养基中明显提高,表明由于寡核苷酸处理,细胞的egf消耗受损。为了证实是消耗的受损(摄入egf至细胞中)而不是细胞的egf表达降低,测量了细胞中的egf的mrna水平,并且清楚地显示出细胞没有表达egf。实施例10培养基中egf浓度的研究如实施例9中看到的,需要培养基中存在一些egf,以在测定法中获得肾毒性读出。这个实施例评价了培养基中的egf浓度对胞内atp水平以及对通过寡核苷酸的靶敲低的作用。在暴露于100μm寡核苷酸连同培养基中的0、1、3、10、30和100ng/mlegf9天后,在汇合的ptec-tert1中测量了胞内atp水平。结果显示于表10中,并且表示三个相同处理的平均。表10:使用培养基中不同的egf水平,作为盐水%的胞内atp为了靶敲低的评价(pcsk9mrna水平),根据制造商的说明,将100μl1×rna裂解混合物(样品处理试剂盒,qs0101)直接加入孔中,从细胞提取了总mrna。将rna裂解混合物保持在-80℃,直至分析。对于基因表达分析,将20μl裂解产物与mrna-捕获磁珠组(panomicsplexset目录号#12697)混合,并孵育过夜,加工用于分支的dna扩增并根据制造商的说明分析(panomicsplex测定法试剂盒,qp1015)。将ppib探针用作管家基因,用于归一化。计算3个生物复制物的平均荧光强度(fi)和标准差,并且相对溶媒对照归一化。结果显示于表11中,并且表示三个相同处理的平均。表11:使用培养基中不同的egf水平,作为盐水%的pcsk9mrna这些结果表明将寡核苷酸施用于ptec-tert1细胞时,对胞内atp水平的作用依赖于培养基中的egf浓度,而靶敲低未受培养基中的egf水平的影响。在培养基中10ng/mlegf下,可以很好地区分寡核苷酸化合物的毒性,而低于3ng/ml时观察到没有显著差异。对于培养基中的egf的需求与实施例6中使用egf作为生物标志物的发现相一致。实施例11.egfr阻断时对测定法的影响这个实施例着手于鉴定阻断用于本发明肾毒性测定法的细胞系上的egfr是否影响测定法。厄洛替尼,egfr激酶活性的小分子抑制剂,以5μm用于这个测定法中。在暴露于100μm寡核苷酸连同含有和不含厄洛替尼的培养基中的10ng/mlegf9天后,测量了汇合的ptec-tert1中的胞内atp水平。结果显示于表12中,并且表示三个相同处理的平均。表12.厄洛替尼存在下对寡核苷酸的胞内atp的作用这些数据表明在使用厄洛替尼灭活egfr的情况中,无害的和有毒的寡核苷酸的atp特征在细胞中是无法区分的。这强烈表明了在基于细胞的测定法中评价寡核苷酸的肾毒性特征时需要egfr表达在细胞中是功能性的。实施例12:寡核苷酸对ptec-tert1细胞中的胞内atp的影响在以上实施例4、6、10和11中,已经显示出评价肾毒性时atp作为egf的替换或补充生物标志物是有用的。研究了来自表1的化合物1-1、3-1和4-1对ptec-tert1细胞中的胞内atp水平的作用需要的条件。按照“材料和方法”部分中所述的培养了ptec-tert1细胞。按照“材料和方法”部分中所述的,用3、10、30或100μm浓度的寡核苷酸处理了汇合的单层ptec-tert1细胞。处理进行了3天或9天。结果显示于以下的表13和14中,并且表示了三个相同处理的平均。表13:单次处理后的atp%表14:3次重复处理后的atp%,其中每次处理使用表中所示的浓度从这可以看出单次处理后第6天,并且特别是第9天,针对严重毒性的化合物4-1可以观察到ptec-tert1细胞中胞内atp的降低。而对于中等毒性的化合物3-1,对于100μm剂量的单次处理和30μm剂量的重复处理,在第9天观察到了对胞内atp的作用。实施例13:寡核苷酸对原代人ptec细胞中的胞内atp的影响基于对人原代ptec细胞和ptec-tert1细胞中的胞内atp水平的体外作用,研究了区分化合物4-1(高毒性)和3-1(中等毒性)之间的毒性程度的能力。按照“材料和方法”部分中所述的培养了细胞。在汇合时,按照“材料和方法”部分中所述的,用下表中所示浓度的寡核苷酸处理了细胞。处理进行了9天。结果显示于以下的表15中,并且表示三个相同处理的平均。表15:剂量应答中的atp%从这可以看出,两种细胞系都能够区分寡核苷酸3-1(中等毒性)和寡核苷酸4-1(高毒性)之间的毒性,同时无毒寡核苷酸1-1没有导致胞内atp的降低。实施例14:环孢菌素a和星形孢菌素对ptec-tert1细胞中的egf水平的作用这个实施例研究了针对以上实施例中的寡核苷酸阐述的体外肾毒性测定法是否也适用于具有已知肾毒性的其他药物化合物。按照“材料和方法”部分中所述的培养了原代petc或ptec-tert1细胞。将原代ptec或ptec-tert1细胞的汇合单层暴露于稀释于dmso中的环孢菌素a(sigma-aldrichc3662)或星形孢菌素(sigma-aldrichs4400),并以最终100μl体积中的1、3、10、30或100μm的终浓度加入细胞培养物中。在处理后3天测量了培养基中的egf的水平。将dmso用作溶媒对照。结果显示于以下的表16中,并且表示三个相同处理的平均。表16:用环孢菌素a或星形孢菌素处理3天,细胞培养物中的egf水平表示为对照%从这些数据,可以看出使用本发明的测定法可以在体外重现两种非常不同的小分子药物环孢菌素a(免疫抑制药物)和星形孢菌素(抗生素药物)的体内肾毒性。数据还表明所述测定法可以检测这些药物降至1μm并且可能甚至更低浓度时的肾毒性。实施例15:寡核苷酸对原代ptec和ptec-tert1细胞中的atp、egf和kim-1的作用化合物20-1,也称为isis,388626,是包含2’-o-甲氧基乙基-rna(moe)修饰的反义寡核苷酸,其靶向钠-葡萄糖共同转运子2(sglt2)。这种化合物已经显示出在13周的研究中每周给药50mg、100mg或200mg能引起人体内可逆转的肾毒性(meer等2016jpharmacolexpthervol359pp280-289)。在这个实施例中,研究了将化合物20-1给予原代ptec细胞或ptec-tert1细胞时通过测量胞外egf和kim-1以及胞内atp是否可以预测这种肾毒性。根据“材料和方法”部分中所述的条件,培养原代ptec细胞和无限增殖化ptec-tert-1细胞系。汇合时,按照“材料和方法”部分中所述的,用30、100、300、400或500μm浓度的寡核苷酸处理细胞。使用“材料和方法”部分中所述的测定法测量了egf、kim-1和atp生物标志物。结果显示于以下的表17和18中,并且表示三个相同处理的平均。表17.原代ptec细胞中寡核苷酸处理后6天的egf和kim-1浓度以及处理9天后的atp浓度表18.ptec-tert1细胞中寡核苷酸处理后6天的egf和kim-1浓度以及处理9天后的atp浓度从这些数据,可以看出使用egf作为生物标志物,使用原代ptec细胞和ptec-tert1细胞都可以预测显示出体内肾毒性的moe反义寡核苷酸(化合物20-1)的体内肾毒性。只在原代ptec细胞中观察到了atp降低,而预测性的kim-1升高只在ptec-tert1细胞中观察到。总之,所述基于egf和atp以及kim-1预测了肾毒性的测定法支持这种预测,这取决于所用的细胞系。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1