一种利用RNA干扰机制的基因调控或编辑系统的制作方法
本发明涉及分子生物学领域,具体而言涉及一种利用rna干扰机制的基因调控或编辑系统。
背景技术
rna干扰机制
rna干扰(rnainterference,rnai)是指由单链或双链rna(double-strandedrna,dsrna)诱发的同源mrna高效特异性降解的现象。rnai具有如下特征:1)rnai是转录后水平的基因沉默机制;2)rnai具有很高的特异性,只降解与之序列相应的单个内源基因的mrna;3)rnai抑制基因表达具有很高的效率,表型可以达到缺失突变体表型的程度,而且相对很少量的dsrna分子(数量远远少于内源mrna的数量)就能完全抑制相应基因的表达,是以催化放大的方式进行的;4)rnai抑制基因表达的效应可以穿过细胞界限,在不同细胞间长距离传递和维持信号甚至传播至整个有机体以及可遗传等特点;5)dsrna不得短于21个碱基,并且长链dsrna也在细胞内被dicer酶切割为21bp左右的sirna,并由sirna来介导mrna切割。而且大于30bp的dsrna不能在哺乳动物中诱导特异的rna干扰,而是细胞非特异性和全面的基因表达受抑和凋亡;6)atp依赖性:在去除atp的样品中rna干扰现象降低或消失显示rna干扰是一个atp依赖的过程。可能是dicer和risc的酶切反应必须由atp提供能量。
小干扰rna(sirna)
病毒基因、人工转入基因、转座子等外源性基因随机整合到宿主细胞基因组内,并利用宿主细胞进行转录时,常产生一些dsrna。宿主细胞对这些dsrna迅即产生反应,其胞质中的核酸内切酶dicer将dsrna切割成多个具有特定长度和结构的小片段rna(大约21~23bp),即sirna(小干扰rna,smallinterferingrna)。sirna在细胞内rna解旋酶的作用下解链成正义链和反义链,继之由反义sirna再与体内一些酶(包括内切酶、外切酶、解旋酶等)结合形成rna诱导的沉默复合物(rna-inducedsilencingcomplex,risc)。risc与外源性基因表达的mrna的同源区进行特异性结合,risc具有核酸酶的功能,在结合部位切割mrna,切割位点即是与sirna中反义链互补结合的两端。被切割后的断裂mrna随即降解,从而诱发宿主细胞针对这些mrna的降解反应。sirna不仅能引导risc切割同源单链mrna,而且可作为引物与靶rna结合并在rna聚合酶(rna-dependentrnapolymerase,rdrp)作用下合成更多新的dsrna,新合成的dsrna再由dicer切割产生大量的次级sirna,从而使rnai的作用进一步放大,最终将靶mrna完全降解。
sirna通常作为rnai的工具被人工合成,但是后来发现很多生物包括线虫还有人类一些特殊的细胞也会合成内源性sirna,以用于基因表达的调控。
微小rna(mirna)
微小rna(microrna,mirna)是一类长度为20-25个核苷酸的非编码小rna,可以在转录后水平调控基因表达。mirbase数据库最新预测结果显示,人类基因组可能表达2656种mirna,这些mirna广泛分布在人体各种组织和器官中。大部分组织和器官都具有专一的mirna表达谱。
掌握各种mirna在不同组织中的表达谱,对于理解相关组织的发育和疾病形成至关重要。2016年nicoleludiwig和他的同事利用芯片技术检测了1997种mirna在两具男性尸体61种组织样本中的表达量。最后他们只检测到1364种mirna至少在其中一种组织中表达,有143种mirna在每种组织中都表达。他们利用组织特异指数tsi(tissuespecificityindex)来定义mirna的分布情况。当某个mirna在每种组织中都表达,那么tsi定义为0,如果这个mirna只在一种组织中特异表达,那么它的tsi则为1。大部分(82.9%)的mirnatsi在分布0.5到0.85之间,表明mirna具有很高的组织特异性。
某些mirna在特定组织中表达量极高。例如,mir-122在成体每个肝脏细胞中的表达量高达66000个分子,是组织中表达量最高的mirna。mir-7和mir-375以及mir-141和mir-200a在脑下垂体中特异表达,mir-142,mir-144,mir-150,mir-155和mir-223在造血细胞中特异表达。mir-144在血管和脾脏中表达量最高,在甲状腺中也有较高表达的量,而在甲状腺乳头癌中降低。另外mir-1-3p,mir-133a-3p,mir-133b在心肌和肌肉中特异表达。这些mirna在肌肉发育中调控关键的基因。mir-338-3p,mir-219-5p,mir-124-3p和mir-9-5p在脑组织中特异表达。mir-507,mir-514a-3p和mir-509-5p只在睾丸中表达。而mir-205-5p只在皮肤中表达,其表达量在生黑色素细胞中最高,随着黑色素瘤的形成下降。
另外,鉴定组织特异表达的mirna可以作为某种疾病在血液中的生物标记物。例如药物导致的肝脏损伤,脂肪肝,乙肝和丙肝的感染以及肝癌都会导致血清中肝脏特异的mir-122表达量的上升。血清中mir-1,mir-206和mir-133a/b的上升,可以作为心脏衰竭以及不同肌肉萎缩症状的生物标记物。另外跑完半程马拉松也会导致血液中这些mirna上升。
各种mirna对基因表达的影响主要在转录后水平。mirna与其靶mrna之间的部分互补导致靶mrna的去稳定化和/或翻译抑制,而mirna与其靶mrna之间的完全或接近完全互补导致靶mrna在特定位置处的切割。许多mirna只在特定组织,细胞类型和发育或疾病阶段表达。因此,mirna谱已被成功用于表征人类肿瘤的发育谱系和分化状态,并且在很多情况下,mirna谱比mrna谱更准确和翔实。另外,在疾病的分化或进展过程中,mirna表达常常动态改变。
综上所述,目前对于mirna或sirna的表达活性与生理状态或疾病的关系是分子生物学领域的研究热点,而同样利用基因调控或编辑的手段进行基因治疗也是医学领域一直关注的研究方向。然而,由于sirna和mirna通常只对基因表达进行抑制性调节,因此可被特定sirna或mirna激活从而对目标基因进行调控或编辑的工具目前仍然是生物技术领域中的空白。
技术实现要素:
本发明人利用mirna介导的sgrna释放策略创建了本发明的micr平台系统,该平台系统可以被特定的内源或外源mirna/sirna激活而启动对目标基因的调控或者编辑。
据此,本发明提供了一种利用rna干扰机制的基因调控或编辑系统,所述基因调控或编辑系统包括以下几个部分:
(1)crispr-cas9基因调控或编辑组件;
(2)前体单链引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列;
所述基因调控或编辑系统可用于对含有目标mirna或sirna的细胞内的目标基因进行控制。
在本发明的一个具体实施方案中,所述目标基因为内源基因或外源基因。
在本发明的另一个具体实施方案中,所述crispr-cas9基因调控或编辑组件为与转录激活因子相连接的核酸酶缺陷型crispr-cas9蛋白。
在本发明的另一个具体实施方案中,所述合成引导rna(sgrna)含有与目标基因的启动子的一段序列互补的序列。
在本发明的另一个具体实施方案中,所述crispr-cas9基因调控或编辑组件为具有核酸酶切割活性的crispr-cas9蛋白或携带碱基修饰酶的核酸酶缺陷型cas9蛋白。
在本发明的另一个具体实施方案中,所述crispr-cas9基因调控或编辑组件为具有基因表达抑制活性的核酸酶缺陷型cas9蛋白。
在本发明的另一个具体实施方案中,所述合成引导rna(sgrna)含有与目标基因的转录起始位点的一段序列互补的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna仅在合成引导rna序列的一端含有与目标mirna或sirna完全互补的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端均含有与目标mirna或sirna完全互补的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端含有的与目标mirna或sirna完全互补的序列是相同或不同的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端含有与一个或多个目标mirna或sirna完全互补的序列。
在本发明的另一方面,还提供了一种利用rna干扰机制的基因调控或编辑方法,所述方法包括以下步骤:
(1)根据将要进行的基因调控或编辑的方式,设计并构建crispr-cas9基因调控或编辑组件;
(2)根据目标基因的目标序列,设计并构建前体单链引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列;
(3)将步骤(1)中构建的crispr-cas9基因调控或编辑组件和步骤(2)中构建的前体合成引导rna引入目标细胞中,如果需要的话,将目标基因也引入目标细胞中。
本发明提供的基因调控或编辑系统通过利用rna干扰机制和crispr-cas9系统相结合,将原本对基因表达起抑制作用的mirna和/或sirna转变为激活本发明的基因调控或编辑系统的信号,从而有效地实现了目标细胞(即表达特定mirna和/或sirna的细胞)内目标基因的调控或编辑。该基因调控或编辑系统不但可以用于表征细胞的状态,更可以用于通过基因调控或编辑手段对特定疾病进行治疗或者改变目标细胞的状态。rna干扰机制与常规基因检测手段相比,具有极高的灵敏度和特异性。因此本发明的系统和方法具有重大的实际意义和广泛的应用前景。
附图说明
参考随附的附图,本发明更多的目的、功能和优点将通过本发明实施方式的如下描述得以阐明,其中:
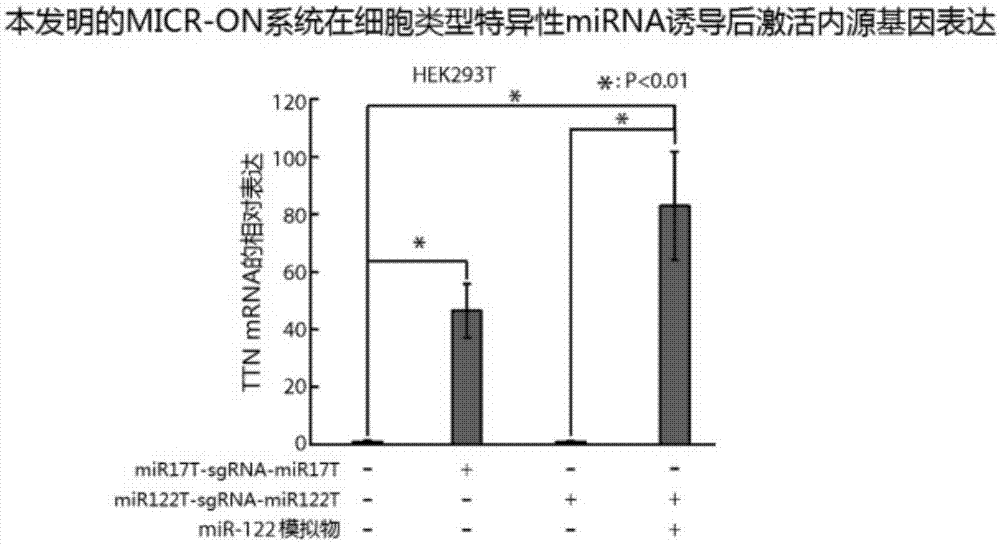
图1为检测本发明的micr-on系统在被细胞特异性mirna或外源mirna和sirna诱导后激活内源基因表达的情况。通过ttn的qrt-pcr进行检测。gapdh基因被用作内参基因。用对照质粒和阴性对照mirna模拟物转染的hek293t细胞的mrna水平作为阴性对照,对数据进行标准化。显示的是平均值±sd,n=3。
图2为检测本发明的micr-be系统在被细胞特异性mirna或外源mirna和sirna诱导后激活内源基因位点编辑的情况。图2a:在基因编辑后用apai消化来自hela细胞的基因组dna的pcr产物的代表性凝胶图像,两个重复实验获得相似的结果。未被切割的条带表示基因编辑的产物,被切割的条带表示未经基因编辑的产物;图2b:对被编辑的目标位点的代表性测序结果,两个重复实验获得相似的结果。当基因组dna中的c被转化为u时,测序结果会由c转变为t。
图3为检测本发明的micr-i系统在被细胞特异性mirna或外源mirna和sirna诱导后抑制内源基因表达的情况。通过dppa5a的qrt-pcr进行检测。gapdh基因被用作内参基因。用对照质粒和阴性对照mirna模拟物转染的v6.5胚胎干细胞的mrna水平作为阴性对照,对数据进行标准化。显示的是平均值±sd,n=3。
具体实施方式
crispr-cas9系统
crispr全称是clusteredregularlyinterspacedshortpalindromicrepeats,中文译作周期间隔短回文重复序列簇,是一类发现于细菌和古细菌中的基因组序列。cas9全称是crispr-associatedprotein9,属于一类核酸酶,cas蛋白最初于2005-2006年左右被鉴定发现。此后几年(2007-2011)的一系列研究逐渐揭示crispr/cas系统作为细菌和古细菌免疫系统对抗病毒侵染的机制:研究发现crispr序列实际上来源于侵入细菌的质粒或者病毒dna,并且crispr序列可以被转录并加工产生短的rna,这些rna片段与cas蛋白结合起到对抗病毒的作用,因此这些rna被称为casrelatedrna,简称crrna。后来又发现细菌需要同时表达另外一种rna来激活cas蛋白的活性,即tracrrna(trans-activationcasrelatedrna,反式激活crrna)。最终在2012年的研究中证明cas9可以和tracrrna、crrna结合,切割质粒dna;研究同时发现tracrrna和crrna可以被串联成一条rna,即sgrna。至此,crispr/cas9系统作为基因调控或编辑工具的条件已经具备,并在2013年被成功应用在哺乳动物的基因编辑上,crispr-cas9时代正式开始。
crispr-cas9系统是目前应用最为广泛的基因编辑系统。这个系统主要由两部分构成:cas9和sgrna。cas9为核酸酶,可以切割dna,造成双链断裂(doublestrandbreak,dsb);sgrna全称为合成引导rna(syntheticguiderna),当cas9和sgrna结合以后,sgrna可以激活并引导cas9蛋白定位到基因组特定位点,由此启动cas9的基因编辑或调控活性。
由于cas9包含两个相对独立的功能区域,结合dna的结构域和切割dna的结构域。将cas9的切割dna活性的结构域突变使其失活后并不会影响cas9结合dna的能力,这种cas9称之为deadcas9,简写为dcas9。而将其他具有基因调控功能的蛋白与dcas9融合表达后可以赋予dcas9相应的基因调控功能。例如,将vp64、p65和rta(简称vpr)3种转录激活因子采用串联的形式与dcas9融合表达后(即dcas9-vp64-p65-rta,简写为dcas9-vpr),可以用与激活与dcas9结合的目标基因的表达水平,而不会造成基因突变。
sgrna:sgrna是一类基于细菌crispr系统、人为自主设计的介导cas9或者dcas9靶向结合目标dna的嵌合rna分子。这类rna含有一个发卡结构,模拟了细菌中tracrrna-crrna复合物结构,用于将cas9或者dcas9蛋白引导至目标基因位点。本发明中使用的是基于酿脓链球菌(streptococcuspyogenes)的改造过的sgrna,包含有三个部分:分别是人为自主设计的长度为20个核苷酸的与目标基因的目标序列互补的部分,长度为40个核苷酸左右的用于与cas9或dcas9结合的序列,以及长度为40个核苷酸左右的细菌来源的稳定区域。由于该合成引导rna(sgrna)含有人为自主设计的与目标基因的目标序列互补配对的序列,从而可以将cas9或dcas9蛋白引导至目标基因的目标序列位置,所以它被称为合成引导rna(sgrna)。在通常的应用中,一般使用u6启动子来表达sgrna。这种启动子通过iii型rna聚合酶转录产生有活性的sgrna。而本发明中使用ii型rna聚合酶的启动子进行转录,那么转录出来的sgrna由于含有5’帽子和3’多聚核苷酸尾巴(polya),不能够发挥功能,因此被称为无活性sgrna。需要通过设计去掉5’帽子和3’polya才能够将其转变为有活性的sgrna。
micr-on平台系统
在本发明的一个方面,本发明人建立了一种可被mirna/sirna诱导的crispr-cas9激活基因表达的实验平台系统(mirna/sirna-induciblecrispr-cas9express-onplatform,简称为micr-on平台),该平台包括以下两个主要部分:1.核酸酶缺陷型crispr-cas9蛋白,即dcas9蛋白;2.前体合成引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列。该平台可以有效地激活含有目标mirna或sirna的特定细胞内的目标基因的表达。
在本发明的一个具体实施方案中,设计了含有与目标基因上游的启动子序列的一段序列互补的sgrna序列,并在该sgrna序列的一端或两端连接与目标mirna或sirna序列完全互补的rna序列,这个就是所述的前体合成引导rna(pre-sgrna,在本发明中也表示为mirt-sgrna-mirt,mirt表示与目标mirna完全互补的序列),在pre-sgrna的两端分别有5’帽子和3’polya,使得该pre-sgrna处于无活性的状态。同时,将dcas9蛋白与转录激活因子相连接以行使其转录活性。在本发明的具体实施方案中,所述目标基因上游的启动子可以是能够驱动目标基因表达的各种启动子。在本发明的一个具体实施方案中,所述转录激活因子为vp64、p65和rta中的一种或多种。
将该平台导入目标细胞后,如果该目标细胞中没有目标mirna的表达,则整个系统处于未被激活的状态,目标基因也不会表达。如果该目标细胞中表达目标mirna或sirna,则该目标mirna或sirna就会与pre-sgrna两端的与其完全互补的rna序列相结合,从而启动rna干扰机制而导致该完全互补rna序列被切割。而该完全互补rna序列的降解则导致‘pre-sgrna两端的5’帽子和3’polya被去掉,从而产生具有活性的sgrna。有活性的sgrna就会将dcas9蛋白引导至目标基因上游的启动子位置,使得dcas9蛋白与该启动子相结合。在结合之后,dcas9蛋白上携带的转录激活因子就驱动了目标基因的表达,从而实现本发明的基因调控的目的。以上过程就是本发明的micr-on平台系统的主要原理。
micr-be平台系统
在本发明的另一个方面,本发明人建立了一种可被mirna/sirna诱导的crispr-cas9激活碱基编辑的实验平台系统(mirna/sirna-induciblecrispr-cas9baseeditingplatform,简称为micr-be平台),该平台包括以下两个主要部分:1.具有核酸酶切割活性的crispr-cas9蛋白或携带碱基修饰酶的ncas9蛋白;2.前体合成引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列。该平台可以有效地对含有目标mirna或sirna的特定细胞内的目标基因的特定位点进行编辑。
在本发明的一个具体实施方案中,设计了含有与目标基因的目标序列互补的sgrna序列,并在该sgrna序列的一端或两端连接与目标mirna或sirna序列完全互补的rna序列,这个就是所述的前体合成引导rna(pre-sgrna,在本发明中也表示为mirt-sgrna-mirt,mirt表示与目标mirna完全互补的序列),在pre-sgrna的两端分别有5’帽子和3’polya,使得该pre-sgrna处于无活性的状态。
将该平台导入目标细胞后,如果该目标细胞中没有目标mirna的表达,则整个系统处于未被激活的状态,目标基因也不会被编辑。如果该目标细胞中表达目标mirna或sirna,则该目标mirna或sirna就会与pre-sgrna两端的与其完全互补的rna序列相结合,从而启动rna干扰机制而导致该完全互补rna序列被切割。而该完全互补rna序列的降解则‘pre-sgrna两端的5’帽子和3’polya被去掉,从而产生具有活性的sgrna。有活性的sgrna就会将具有核酸酶切割活性的crispr-cas9蛋白或携带碱基修饰酶的dcas9蛋白引导至目标基因的目标序列位置,使它们互相结合。在结合之后,目标基因被具有核酸酶切割活性的crispr-cas9蛋白切割,或者被dcas9蛋白携带的碱基修饰酶修饰,从而实现本发明的基因编辑的目的。以上过程就是本发明的micr-be平台系统的主要原理。
micr-i平台系统
在本发明的另一个方面,本发明人建立了一种可被mirna/sirna诱导的crispr-cas9激活基因抑制的实验平台系统(mirna/sirna-induciblecrispr-cas9inhibitingplatform,简称为micr-i平台),该平台包括以下两个主要部分:1.具有基因表达抑制活性的dcas9蛋白;2.前体合成引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列。该平台可以有效地对含有目标mirna或sirna的特定细胞内的目标基因的转录进行抑制。
在本发明的一个具体实施方案中,设计了含有与目标基因的目标序列互补的sgrna序列,并在该sgrna序列的一端或两端连接与目标mirna或sirna序列完全互补的rna序列,这个就是所述的前体合成引导rna(pre-sgrna,在本发明中也表示为mirt-sgrna-mirt,mirt表示与目标mirna完全互补的序列),在pre-sgrna的两端分别有5’帽子和3’polya,使得该pre-sgrna处于无活性的状态。在本发明的另一个具体实施方案中,所述合成引导rna(sgrna)含有与目标基因的转录起始位点的一段序列互补的序列。
将该平台导入目标细胞后,如果该目标细胞中没有目标mirna的表达,则整个系统处于未被激活的状态,目标基因也不会被抑制。如果该目标细胞中表达目标mirna或sirna,则该目标mirna或sirna就会与pre-sgrna两端的与其完全互补的rna序列相结合,从而启动rna干扰机制而导致该完全互补rna序列被切割。而该完全互补rna序列的降解则导致‘pre-sgrna两端的5’帽子和3’polya被去掉,从而产生具有活性的sgrna。有活性的sgrna就会将具有基因抑制活性的dcas9蛋白引导至目标基因的目标序列位置,使它们互相结合。在结合之后,目标基因被dcas9蛋白携带的转录抑制因子所抑制,从而实现本发明的基因调控的目的。以上过程就是本发明的micr-i平台系统的主要原理。
在本发明的具体实施方案中,所述目标基因为内源基因或外源基因。所述外源基因可以是人为引入的基因。在具体的实施方案中,可以在基因组中引入外源基因,并使得该外源基因受到本发明的基因调控或编辑系统的控制,仅在特定的条件下(例如存在目标mirna或sirna的条件下)被激活或抑制。
在本发明的另一个具体实施方案中,所述前体合成引导rna仅在合成引导rna序列的一端含有与目标mirna或sirna完全互补的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端均含有与目标mirna或sirna完全互补的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端含有的与目标mirna或sirna完全互补的序列是相同或不同的序列。
在本发明的另一个具体实施方案中,所述前体合成引导rna在合成引导rna的序列两端含有与一个或多个目标mirna或sirna完全互补的序列,这样使得本发明的基因调控或编辑系统可以被一个或多个目标mirna或sirna信号所激活。
在本发明的另一方面,还提供了一种利用rna干扰机制的基因调控或编辑方法,所述方法包括以下步骤:
(1)根据将要进行的基因调控或编辑的方式,设计并构建crispr-cas9基因调控或编辑组件;
(2)根据目标基因的目标序列,设计并构建前体单链引导rna(pre-sgrna),所述前体合成引导rna含有与目标mirna或sirna完全互补的序列以及合成引导rna(sgrna)的序列;
(3)将步骤(1)中构建的crispr-cas9基因调控或编辑组件和步骤(2)中构建的前体合成引导rna引入目标细胞中,如果需要的话,将目标基因也引入目标细胞中。
本发明提供的基因调控或编辑系统通过利用rna干扰机制和crispr-cas9系统相结合,将原本对基因表达起抑制作用的mirna和/或sirna转变为激活本发明的基因调控或编辑系统的信号,从而有效地实现了目标细胞(即表达特定mirna和/或sirna的细胞)内目标基因的调控或编辑。该基因调控或编辑系统不但可以用于表征细胞的状态,更可以用于通过基因调控或编辑手段对特定疾病进行治疗或者改变目标细胞的状态。rna干扰机制与常规基因检测手段相比,具有极高的灵敏度和特异性。因此本发明的系统和方法具有重大的实际意义和广泛的应用前景。
术语和缩写
在本说明书中使用了一些术语和缩写,其含义如下所述,未特别说明的术语和缩写具有本领域技术人员公知的含义。
rnai:rna干扰(rnainterference);
sirna:小干扰rna(smallinterferingrna);
mirna:微小rna(microrna);
mir:各种mirna的缩写,通常后面跟有数字及字母编号以表示其命名,各种mir的编号及其序列在本领域中都是公知的;
mirt:表示与目标mirna或sirna完全互补的序列(mirnatarget);
sgrna:合成引导rna(syntheticguiderna);
pre-sgrna:前体合成引导rna,含有与目标mirna或sirna完全互补的序列以及sgrna的序列,亦可表示为mirt-sgrna或者mirt-sgrna-mirt;
crispr:周期间隔短回文重复序列簇(clusteredregularlyinterspacedshortpalindromicrepeats);
cas9:周期间隔短回文重复序列簇相关蛋白9(crispr-associatedprotein9);
dcas9:核酸酶缺陷型cas9,由于其核酸酶活性区域被突变,从而丧失了dna切割活性而仅保留了dna结合活性;
ncas9:cas9切口酶,是cas9另一种突变体,其第10位的天冬氨酸(d)突变为丙氨酸(a)后,只能切割与非互补的dna链,从而提高编辑效率。micr-on:可被mirna/sirna诱导的crispr-cas9激活基因表达的平台系统(mirna/sirna-induciblecrispr-cas9express-onplatform);
micr-be:可被mirna/sirna诱导的crispr-cas9激活碱基编辑的平台系统(mirna/sirna-induciblecrispr-cas9baseeditingplatform);
micr-i:可被mirna/sirna诱导的crispr-cas9激活基因抑制的平台系统(mirna/sirna-induciblecrispr-cas9inhibitingplatform);
dcas9-vpr:将vp64、p65和rta(简称vpr)3种转录激活因子采用串联的形式与dcas9连接的产物;
rapobec:大鼠胞嘧啶脱氨酶apolipoproteinbmrnaeditingenzyme,catalyticpolypeptide。
ugi:尿嘧啶dna糖基酶活性抑制剂,来源于枯草芽孢杆菌噬菌体pbs1的83个残基,可以帮助提高基因编辑(胞嘧啶转变胸腺嘧啶)的活性。
通过下面的参考示范性实施例以及附图,本发明的目的和功能以及用于实现这些目的和功能的方法将得以阐明。然而,本发明并不受限于以下所公开的示范性实施例;可以通过不同形式来对其加以实现。说明书的实质仅仅是帮助相关领域技术人员综合理解本发明的具体细节。
实施例1
材料和方法
质粒和载体的构建
将dcas9-vpr连接于含有由pgk启动子驱动的潮霉素抗性基因的piggybac载体中,所述dcas9-vpr由caggs启动子驱动。将pre-sgrna(两端含有与目标mirna或sirna完全互补的rna序列的sgrna序列,简称为mirt-sgrna-mirt)连接于含有由pgk启动子驱动的博来霉素(zeocin)抗性基因的piggybac载体中,所述pre-sgrna也由caggs启动子驱动。将由具有tre3g元件的上游cmv小启动子控制的红色荧光蛋白(rfp)连接于具有杀稻瘟菌素抗性基因的piggybac载体中。
其他两种质粒(rapobec-ncas9-ugi(be3))和dcas9-krab的制备。将ncas9-apobec-ugi(be3)连接于含有由pgk启动子驱动的潮霉素抗性基因的piggybac载体中,所述ncas9-apoec-ugi(be3)由caggs启动子驱动。将dcas9-krab连接于含有由pgk启动子驱动的潮霉素抗性基因的piggybac载体中,所述dcas9-krab由caggs启动子驱动。
为了制备对目标mirna或sirna敏感的mirt-sgrna-mirt构建体,使用fastpfu聚合酶(transgene公司),以含有所需sgrna的质粒为模板,用携带mirt序列的引物进行pcr。pcr产物与ecori-hf(20u,neb公司)和bamhi-hf(20u,neb公司)在37℃下孵育2h,用旋转柱(magen公司)纯化,并用t4连接酶(lifetechnology公司)连接到经过ecori和bamhi消化的piggybac载体上。
细胞培养,质粒转染和小rna转染
如前人所报道的方法,在明胶或辐照的小鼠成纤维细胞上培养野生型的和dgcr8-/-胚胎干细胞(esc)。在添加有10%fbs(pansera公司)的高糖dmem培养基(gibco公司)上,以5%co2和37℃的条件培养hek293t细胞和hela细胞。
对于激活内源基因表达实验,将hek293t细胞以50,000个细胞/孔的密度接种于聚-d-赖氨酸(sigma-aldrich公司)包被的48孔板中。18小时后,用20nm终浓度的mir-122模拟物或阴性对照物转染细胞。6小时后,使用fugenehd(promega)转染试剂按照每孔125ngdcas9-vpr和125ngpre-sgrna的质粒剂量转染细胞,并同时用空质粒转染细胞设为阴性对照。转染48小时后,收集细胞并用trizol试剂盒提取总rna,用于mrna的qrt-pcr。
对于碱基编辑实验,将hela细胞以50,000个细胞/孔的密度接种于24孔板(sigma-aldrich公司)中。18小时后,用20nm终浓度的mir-122模拟物或阴性对照物转染细胞。6小时后,用含有250ngapobec-ncas9-ugi(be3)和250ngpre-sgrna的质粒转染细胞,并同时用空质粒转染细胞设为阴性对照。转染48小时后,用10μg/ml杀稻瘟菌素s(gibco公司),500μg/ml潮霉素(roche公司)处理细胞3天。收集细胞并提取基因组dna以基因组为模板,用特定引物pcr扩增目的条带。利用胶回收试剂盒(magen公司)回收目的片段后,用apai(neb公司)酶切鉴定碱基编辑效率。
对于抑制基因表达实验,将小鼠胚胎干细胞v6.5以50,000个细胞/孔的密度接种于0.25%明胶包被的12-well板(sigma-aldrich公司)中。18小时后,使用lipofectamine3000转染试剂盒(lifetechnology公司)将上述制备的含有dcas9-krab和pre-sgrna的质粒与pbase表达质粒一起共转染胚胎干细胞。24小时后,用10μg/ml杀稻瘟菌素s(gibco公司),150μg/ml潮霉素(roche公司)和100μg/ml博来霉素(invitrogen公司)处理细胞4天。之后,铺单细胞于照射过的小鼠滋养层细胞上。6天后,挑克隆。然后将合适的克隆以10,000个细胞/孔的密度接种于0.25%明胶包被的12-well板(sigma-aldrich公司)中。18小时后,用20nm终浓度的mir-122模拟物或阴性对照物转染细胞,收集细胞并用trizol试剂盒提取总rna,用于mrna的qrt-pcr。
qrt-pcr和mirnaqrt-pcr
使用trizol试剂盒(roche公司)提取rna,并用biodropsisbd2000超微量核酸蛋白分析仪进行定量。将500ng左右的rna使用第一cdna合成试剂盒(vazyme公司)进行反转录。用abisteponeplus实时荧光定量pcr系统(appliedbiosystems公司)进行定量pcr。对于mirnaqpcr,根据生产商的说明书,使用vazyme公司的mirna第一链cdna合成试剂盒(通过茎环)反转录rna样品。使用mirnauniversalsybrqpcrmastermix系统(vazyme公司)进行qpcr反应。使用v6.5胚胎干细胞作为对照,基于qpcr结果计算mirna拷贝数。根据前人报道的方法,v6.5胚胎干细胞中的mir-294的拷贝数估算为每细胞2339拷贝。
统计学分析
除非另有说明,数据以平均值±sd表示。我们进行双尾不成对student’st检验以确定统计学显著性。p值<0.05被认为是具有统计学显著性。
实施例2
本发明的micr-on系统在细胞类型特异性mirna诱导后激活内源基因表达
在本实施例中,我们测试了本发明的micr-on系统是否可以由细胞类型特异性mirna触发以激活内源基因的转录。我们构建了dcas9蛋白和vp64、p65和rta这3种转录激活因子串联的融合蛋白(简称dcas9-vpr),作为本实施例的crispr-cas9基因调控或编辑组件。然后,我们设计了两种pre-sgrna,分别为mir17t-sgrna-mir17t和mir122t-sgrna-mir122t,其中的sgrna均靶向于人类肌联蛋白(titin,ttn)的转录起始位点(transcriptionalstartsite,tss)。然后,我们将表达dcas9-vpr和这两种pre-sgrna的质粒分别转染到hek293t细胞中,该细胞表达高水平的mir-17但不表达mir-122。结果示于图1,如图所示,在含有dcas9-vpr和mir17t-sgrna-mir17t的细胞中ttn的表达显著增加,而在含有dcas9-vpr和mir122t-sgrna-mir122t的细胞中或不含有任何pre-sgrna的细胞(阴性对照)中没有ttn的表达;此外,将mir-122模拟物转染到含有dcas9-vpr和mir122t-sgrna-mir122t的hek293t细胞以后,可导致ttn转录的显著增加。以上数据表明,本发明的micr-on系统可被细胞类型特异性mirna诱导后激活内源基因表达。
实施例3
本发明的micr-be系统在细胞类型特异性mirna的诱导后激活基因组dna的碱基编辑
在本实施例中,我们测试了本发明的micr-be系统在细胞类型特异性mirna的诱导后激活基因组dna的碱基编辑的效果。根据前人报道的方法,我们构建了表达apobec-ncas9-ugi(也称为be3)的质粒(参见komor,a.c.,kim,y.b.,packer,m.s.,zuris,j.a.&liu,d.r.programmableeditingofatargetbaseingenomicdnawithoutdouble-strandeddnacleavage.nature533,420-424(2016)),作为本实施例的crispr-cas9基因调控或编辑组件。rapobec-ncas9-ugi(be3)是一种融合蛋白构建体,其表达产物为携带有大鼠胞嘧啶脱氨酶和尿嘧啶dna糖基酶活性抑制剂的ncas9融合蛋白。此外,我们设计了两种pre-sgrna,分别为mir21t-sgrna-mir21t和mir294t-sgrna-mir294t,其中的sgrna靶向于linc01509基因的最后一个内含子,并将表达be3的质粒和这两种pre-sgrna的质粒分别转染到hela细胞中。hela细胞表达高水平的mir-21,但不表达mir-294。结果示于图2,如图所示,在含有be3和mir21t-sgrna-mir21t的细胞的基因组dna预期位置观察到从c转化为u的高百分比转化率,在含有be3和mir294t-sgrna-mir294t的细胞或不含任何pre-sgrna的细胞(阴性对照)中没有从c到u的转化。此外,将mir-294模拟物转染到含有be3和mir294t-sgrna-mir294t的细胞以后,我们观察到在细胞的基因组dna预期位置从c至u的转化率约为40%。综上所述,以上数据表明本发明的micr-be系统可以通过细胞类型特异性mirna的诱导实现对基因组dna的高水平碱基编辑。
实施例4
本发明的micr-i系统在细胞类型特异性mirna的诱导后抑制目标基因的表达
在本实施例中,我们测试了本发明的micr-i系统是否可以由细胞类型特异性mirna触发以抑制内源基因的转录。我们首先构建了dcas9蛋白与krab锌指蛋白的融合蛋白(简称dcas9-krab)作为本实施例的crispr-cas9基因调控或编辑组件,krab锌指蛋白是常用的转录抑制因子。然后,我们设计了两种pre-sgrna,分别为mir294t-sgrna-mir294t和mir122t-sgrna-mir122t,其中的sgrna均靶向于内源基因dppa5a的转录起始位点(transcriptionalstartsite,tss),该基因是一个在胚胎干细胞中高表达的基因。然后,我们将表达dcas9-krab和这两种pre-sgrna的质粒分别转染到胚胎干细胞中,该细胞表达高水平的mir-294但不表达mir-122。结果示于图3,如图所示,在含有dcas9-krab和mir294t-sgrna-mir294t的细胞中dppa5a的表达显著被抑制,而在含有dcas9-krab和mir122t-sgrna-mir122t的细胞中或不含有任何pre-sgrna的细胞(阴性对照)中dppa5a的表达显著未被抑制;此外,将mir-122模拟物转染到含有dcas9-krab和mir122t-sgrna-mir122t的v6.5胚胎干细胞以后,可导致dppa5a的表达显著被抑制。以上数据表明,本发明的micr-i系统可被细胞类型特异性mirna诱导后抑制内源基因表达。
通过公开的上述实施例,本发明的其他实施例对于本领域技术人员都是易于想到和理解的。说明书中的具体描述仅被认为是示例性的,本发明的真正范围和主旨均由本发明的权利要求书所限定。
- 还没有人留言评论。精彩留言会获得点赞!