一种D-泛酸钠合成和手性拆分方法与流程
本发明涉及化学合成领域,更具体的说是涉及一种d-泛酸钠合成和手性拆分方法。
背景技术:
泛酸是b族维生素类物质,为水溶性维生素的一种,广泛存在于生物界,俗称维生素b5,于1933年首次从酵母中分离获得,可分为dl-型(混旋体),d-型(右旋体),l-型(左旋体),只有d-型体(右旋体)具有生物活性,d-泛酸钠是泛酸d-型对映异构体的钠盐。
泛酸在体内主要以辅酶a形式参与多种代谢环节,包括碳水化合物、蛋白质、脂肪及上皮功能维持正常所必需的辅酶a的组成部分。临床上用于泛酸缺乏(如吸收不良综合症、热带口炎性腹泻、乳糜泻、局限性肠炎或泛酸拮抗药物应用)的预防和治疗。
常用泛酸为钙盐或钠盐,广泛应用于医药、饲料、食品等行业中,药用泛酸通常与其他b族维生素制成复合制剂,泛酸钙及其片剂已经收载于2015版国家药典。由于泛酸经常需要与其他b族维生素如核黄素磷酸钠合用,会形成磷酸钙降低药物效价而产生配伍禁忌,采用泛酸钠则可避免,达到安全有效用药的目的。因此,泛酸钠在药用价值上更优于泛酸钙,符合安全有效的药品使用要求。
d-泛酸(钙盐或钠盐)的合成文献报道的有生物法和化学法。
中国专利cn1946851公开了一种从发酵培养液制备d-泛酸钙的方法,采用的产d-泛酸钙微生物种类、培育方法和纯化用强碱性阴离子树脂均是公知的,或者商品化的,优点在于该方法的适应性强,适合规模化生产。但发酵法制备的泛酸钠含量低,有关物质高,一般用于食品和饲料添加,并且存在工艺稳定性差,生产周期长,受温度和ph等条件影响显著等问题,更难以达到药用要求。
中国专利cn101948402公开了采用b-氨基丙腈经碱水解后与d-泛解酸内酯酰化,再用钙离子树脂纯化成钙盐的方法。该方法不采用氧化钙或氢氧化钙作为钙化剂,避免了成钙盐过程中生成水对酰化反应的影响,收率可达到90%。但该工艺存在工艺条件苛刻,生成时间长,采用大量毒性试剂等问题,并且工艺中产生大量金属盐类废弃物,加重了环境污染的风险。另外,工艺中采用强碱强热工艺,存在d-泛酸消旋的风险。
中国专利cn103045579公开了一种泛酸钠的制备方法。以市售泛酸钠原料,以乙醇、乙醚为结晶溶剂,结合超声进行重结晶。优点在于工艺简单,增加了泛酸钠的稳定性。但是该工艺只属于泛酸钠的精制工艺,原料带入的有关物质难以有效去除。另外,在实际运用中,发现泛酸钠水中溶解度较高,按此工艺收率较低,再现性不强,难以推广应用。
目前,泛酸钙的工艺较为成熟,国内外公开或报道了大量的制备方案,但是泛酸钠的制备报道较少,尤其是药用泛酸钠,多以泛酸钙离子交换成钠盐或市售泛酸钠精制工艺。一方面,市售泛酸钙多为食品级或饲料级,简单的离子交换或重结晶工艺难以有效去除原料带入的有关物质,尤其是泛酸钙合成过程中的水解副产物,与泛酸结构类似,上述工艺难以去除。工艺路线较短,将泛酸钠生产的质量控制放在结晶工序,增加了质量偏差发生的风险。泛酸酰化工艺中采取的强碱条件,存在消旋风险,造成异构体杂质升高而药效降低。因此,原有工艺的缺陷难以使泛酸钠达到注射剂的药用要求。另一方面泛酸钠合成工艺中,存在大量氰类和金属盐废水以及氨等废气,此类废水难以采用生化处理,造成巨大的环境污染压力。因此,需要提出新的合成或技术方案,根据原料药工艺中起始物料的选择和杂质谱研究,控制泛酸合成中水解产物,将有关物质的控制放在泛酸合成过程中,并采用合适的,经济的方法进行拆分,达到药用泛酸钠的质量标准和稳定性要求。
技术实现要素:
本发明要解决的技术问题是克服现有技术中泛酸钠的生产泛酸合成工艺中存在大量氰类和金属盐废水以及氨等废气,此类废水难以采用生化处理,造成巨大的环境污染压力的缺陷,提供一种d-泛酸钠合成和手性拆分方法。
为了解决上述技术问题,本发明提供了如下的技术方案:
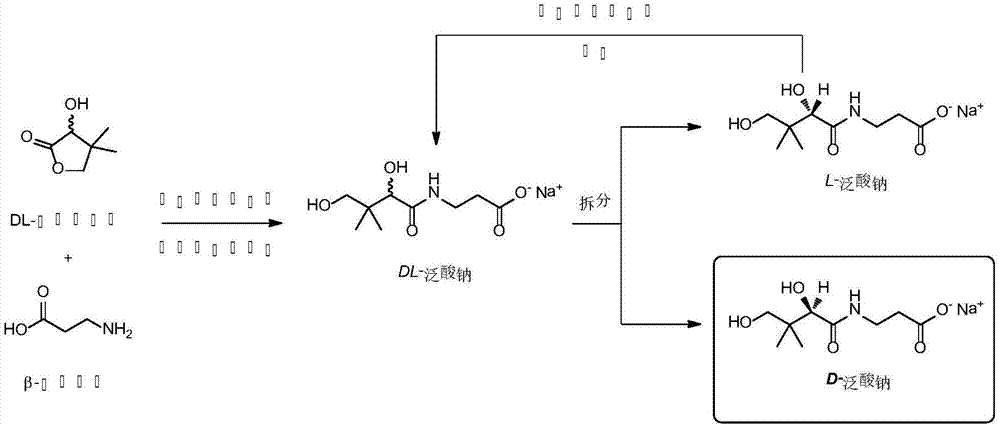
一种d-泛酸钠合成和手性拆分方法,包括以下步骤:
步骤1:向β-氨基丙酸的甲醇或乙醇中加入有机碱,搅拌溶解至澄清;甲醇或乙醇,都为无水溶剂,优选甲醇。有机碱为甲醇钠、乙醇钠或由金属钠与甲醇预制。
步骤2:在-10~10℃下,加入dl-泛解酸内酯,搅拌反应2~8小时,反应完毕后过滤;
步骤3:滤液用冰醋酸调节ph后,加热减压浓缩至相对比重0.94-0.99后冷却至室温,搅拌结晶12h;
步骤4:结晶产物加2-5倍体积的甲醇或乙醇溶液回流溶解,再加0.6~1.5%活性炭搅拌15min,趁热过滤后向滤液加入l-泛酸钠(以结晶产物重量计),结晶12小时后过滤,得l-泛酸钠粗品;l-泛酸钠可用于下次生产诱导结晶,也可溶于无水甲醇或乙醇,用甲醇钠或乙醇钠消旋后再诱导结晶。
步骤5:滤液减压浓缩至相对比重0.94-0.99后冷却至室温,加入d-泛酸钠晶种,结晶12h后过滤,得d-泛酸钠,经过若干次诱导结晶,直至获得旋光度检测符合规定的结晶产品;
步骤6:获得d-泛酸钠在40~50℃下,减压干燥4小时,即得。
进一步的,所述步骤1)中β-氨基丙酸与甲醇钠或乙醇钠摩尔比为1:1.1~1.5,所述步骤2)中β-氨基丙酸与dl-泛解酸内酯摩尔比为1:1~1.1。
进一步的,所述步骤3)冰醋酸调节ph至9.2-10.2,减压浓缩温度应控制在45~55℃。
进一步的,将所述步骤4)所得的l-泛酸钠粗品用2~5倍体积的5~10%甲醇或乙醇回流溶解,然后加入1~3倍摩尔比的甲醇钠或乙醇钠,反应2h后得到消旋的dl-泛酸钠溶液,可与下批产品一起诱导拆分。
进一步的,所述的甲醇或乙醇溶液都为含水溶液,其中,甲醇溶液含水2.5~5.5%,乙醇溶液含水3.0~7.5%。
进一步的,所述步骤3)和步骤5)中减压浓缩后溶液相对比重的检测是在溶液40~50℃条件下进行的。
进一步的,步骤4和步骤5中加入的d-泛酸钠晶种的用量为1~5%(以待拆分的混旋或低旋光性产品重量计),诱导拆分温度应<30℃。
本发明所达到的有益效果是:本发明所述d-泛酸钠合成和拆分方法,采用市售易得价廉的原料,经一步酰化反应,一步诱导拆分,获得高光学纯度的d-泛酸钠,不需用手性原料或手性拆分试剂,反应条件温和,时间短,收率高。其中,d-泛酸钠经一步诱导结晶旋光值可到到+25°以上,经二步诱导结晶可达到+26°以上。以β-氨基丙酸计,最终收率可达到45%以上。一步诱导结晶副产物l-泛酸钠经消旋后回用至后续生产,平均收率达到60%以上。
本发明提供了一种d-泛酸钠合成和手性拆分方法,能够解决生物法生产周期长,纯度低以及化学法的质量控制风险和环境污染问题,进而达到d-泛酸钠作为注射剂安全有效的药用要求。该方法首先采用dl-泛解酸内酯与b-氨基丙酸酰化后,诱导结晶拆分获得d-泛酸钠。β-氨基丙酸与dl-泛解酸内酯摩尔比为1:1~1.1,加入的诱导用晶种的用量为1~5%。本发明的合成方法,未采用毒性试剂或大量金属盐,有机溶剂都可回收处理后再利用,产生的少量含钠盐废水可经简单的离子交换进行水处理。彻底解决了传统生物法和化学法的环境污染问题。
附图说明
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
图1是本发明的反应方程式;
图2是1h-nmr(400mhz,dmso-d6)图谱;
图3是13c-nmr(400mhz,dmso-d6)图谱;
图4是13c-dept135°(400mhz,dmso-d6)图谱;
图5是ms:[m+h]+=242;[m-na]+=218(exactms:241)图谱。
具体实施方式
以下对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
实施例1
将100gβ-氨基丙酸加入到800ml无水甲醇中,分批加入60.97g甲醇钠。搅拌溶解至澄清。降温至-10℃,加入dl-泛解酸内酯146.1g,搅拌反应4h。反应结束经0.8mm过滤器过滤。滤液用10%乙酸的甲醇溶液调节ph至9.2,50℃减压浓缩至相对比重为0.966,降至室温后结晶12h,得dl-泛酸钠粗品241g。dl-泛酸钠粗品用500ml的3%甲醇水溶液溶解,回流溶解后加5g活性炭搅拌15min,趁热过滤。滤液冷却到30℃时加入5gl-泛酸钠,再冷却至室温后结晶12小时后过滤,得l-泛酸钠84.24g。滤液50℃下减压浓缩至相对比重为0.947,冷却到30℃时加入5gd-泛酸钠,再冷却至室温后结晶12小时后过滤,得d-泛酸钠129.89g,收率48.11%(以b-氨基丙酸计),旋光度:
实施例2
将100gβ-氨基丙酸加入到800ml无水甲醇中,分批加入72.98g甲醇钠。搅拌溶解至澄清。降温至-10℃,加入dl-泛解酸内酯175.3g,搅拌反应4h。反应结束后加入上批消旋获得的dl-泛酸钠甲醇溶液,混合液经0.8mm过滤器过滤。滤液用10%乙酸的甲醇溶液调节ph至9.5,50℃减压浓缩至相对比重为0.965,降至室温后结晶12h,得dl-泛酸钠粗品315.27g。dl-泛酸钠粗品用600ml的4%乙醇水溶液溶解,回流溶解后加6g活性炭搅拌15min,趁热过滤。滤液冷却到30℃时加入6gl-泛酸钠,再冷却至室温后结晶12小时后过滤,得l-泛酸钠粗品119.85g。滤液50℃下减压浓缩至相对比重为0.977,冷却到30℃时加入5gd-泛酸钠,再冷却至室温后结晶12小时后过滤,得d-泛酸钠156.34g,收率57.90%(以β-氨基丙酸计),旋光度:
实施例3
将100gβ-氨基丙酸加入到1000ml无水乙醇中,分批加入114.68g乙醇钠。搅拌溶解至澄清。降温至-5℃,加入dl-泛解酸内酯163.96g,搅拌反应8h。反应结束后加入上批消旋获得的dl-泛酸钠乙醇溶液,混合液经0.8mm过滤器过滤。滤液用10%乙酸的乙醇溶液调节ph至10.0,50℃减压浓缩至相对比重为0.982,降至室温后结晶12h,得dl-泛酸钠粗品335.77g。dl-泛酸钠粗品用650ml的5%乙醇水溶液溶解,回流溶解后加6g活性炭搅拌15min,趁热过滤。滤液冷却到30℃时加入6gl-泛酸钠,再冷却至室温后结晶12小时后过滤,得l-泛酸钠粗品115.45g。滤液50℃下减压浓缩至相对比重为0.973,冷却到30℃时加入5gd-泛酸钠,再冷却至室温后结晶12小时后过滤,得d-泛酸钠186.14g,收率68.94%(以b-氨基丙酸计),旋光度:
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!