一种2-甲基丝氨酸的制备方法与流程
本发明涉及一种丝氨酸的制备方法,特别涉及一种2-甲基丝氨酸(cas:5424-29-3)的制备方法。
背景技术:
丝氨酸是又名β-羟基丙氨酸,是一种天然存在的氨基酸,它在脂肪和脂肪酸的新陈代谢及肌肉的生长,免疫血球素和抗体的产生,在细胞膜的制造加工,包围神经细胞的鞘的合成中都发挥着作用。对丝氨酸进行结构修饰而产生的2-甲基丝氨酸是药物研究、多肽合成中非常重要的一种非天然氨基酸,无法通过自然界的提取而相对难以获得。但它的需求随着科学研究的深入而越来越大,所以开发一种简单快捷的化学合成方法来生产2-甲基丝氨酸是有重大的科学研究及经济意义的。
目前合成α-甲基-l-丝氨酸的方法:
1)tetrahedronletts(四面体快报),1998,39,3749
2)tetrahedronasymmetry(不对称四面体),1998,9,1703
3)org.lett(科学引文索引),2003,5,1035
4)专利cn20170009528报道了一种利用手性磷酸的合成方法
在现有的文献报道中合成2-甲基丝氨酸大多利用贵金属催化的方法通过不对称双羟基化构建关键的手性中心,再经历多步反应得到最终目标产物。路线长,操作复杂,不利于大规模生产。本发明主要解决的时利用很便宜的大规模工业生产丙氨酸作为手性控制源,再通过简单的几步化学反应最终得到高化学纯度高手性纯度的甲基丝氨酸。
技术实现要素:
本发明的目的在于提供一种2-甲基丝氨酸的制备方法,主要解决现有合成方法路线长,操作复杂,不利于大规模生产的技术问题。利用化学合成方法合成2-甲基丝氨酸是大规模获得该非天然氨基酸的重要方法,而其中最关键的是寻找便宜且能大规模工业生产手性控制源。经长期研究,发现由手性丙氨酸经过简单反应后生产的手性辅剂1完全符合我们要求的手性控制源,利用它经过三步常规反应我们就得到了高化学纯度高手性纯度的2-甲基丝氨酸。其反应路线如下:
本发明的技术方案如下:一种2-甲基丝氨酸的制备方法,其特征在于:包括以下步骤:
(1)cbz-手性丙氨酸与苯甲醛二甲缩醛在氯化亚砜和氯化锌的作用下反应,反应产物经结晶得到中间体1;
(2)中间体1与氯甲基苄醚在有机金属碱性试剂的配合下反应得到中间体2;
(3)中间体2在无机碱的作用下得到中间体3;
(4)中间体3溶于溶剂最后经过在钯炭催化氢解得到最终产物2-甲基丝氨酸。
具体按照如下步骤:
(1):将cbz-手性丙氨酸溶于四氢呋喃中,加入苯甲醛二甲缩醛,通入氮气置换体系,冷冻盐水,滴入氯化亚砜,滴完,分批加入无水氯化锌控制温度低于0℃,搅拌3~6小时,加入水淬灭反应,然后用乙酸乙酯萃取,洗涤,干燥,浓缩,用异丙醚和石油醚混合溶剂重结晶,过滤,烘干得到中间体1,为白色固体;
(2):将中间体1溶于四氢呋喃中,通入氮气置换体系,液氮降温至-78℃,滴入六甲基二硅基胺基锂,低温搅拌反应,滴入氯甲基苄醚,滴完继续搅拌,然后自然升温至20~25℃,冷水淬灭反应,再加入乙酸乙酯萃取,有机相再用水洗涤一次,干燥,旋干,得到粗品中间体2,为淡黄色油状物,直接下一步;
(3):将粗品中间体2溶于四氢呋喃,加入氢氧化锂溶液,室温搅拌,用乙酸乙酯洗涤,水层用盐酸调ph至3~4左右,加入乙酸乙酯萃取水相,乙酸乙酯相再用水洗涤,干燥,过滤,浓缩得中间体3,为棕色油状物;
(4):将中间体3溶于溶剂,加入钯炭,通入氢气升温反应5~6小时,过滤,固体用水洗涤,滤液和洗液浓缩得粗品,粗品用丙酮打浆,过滤,烘干,得到最终产物2-甲基丝氨酸,为白色固体。
步骤(1)所用的手性丙氨酸可以是d-丙氨酸或l-丙氨酸,用d-丙氨酸得到的是2-甲基-l-丝氨酸,用l-丙氨酸得到的是2-甲基-d-丝氨酸。
步骤(1)所用的氯化锌为无水氯化锌,所用量为1~1.2eq,优选1.1eq。
步骤(1)所用氯化亚砜量为1~1.5eq,优选1.2eq。
步骤(2)所用的有机金属碱性试剂选自六甲基二硅基胺基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或二异丙基氨基锂中的一种,优选六甲基二硅基胺基锂。
步骤(2)反应温度为-76℃~-40℃,优选-65℃~-50℃。
步骤(3)所用的无机碱选自氢氧化锂、氢氧化钠或氢氧化钾中的一种,优选氢氧化锂。
步骤(4)所用的溶剂为甲醇、乙醇、四氢呋喃或水中的一种或者任意两种的混合液,优选甲醇与水的混合液。
步骤(4)反应温度20℃~80℃,优选60℃。
步骤(4)反应压力0.1mpa~4mpa,优选3mpa。
本发明的有益效果是:本发明用一种工业大量使用的成本低廉丙氨酸为手性控制源,来立体选择性合成2-甲基丝氨酸。所用原料成本低,步骤短,工艺简单,适合工业化大量生产。
附图说明
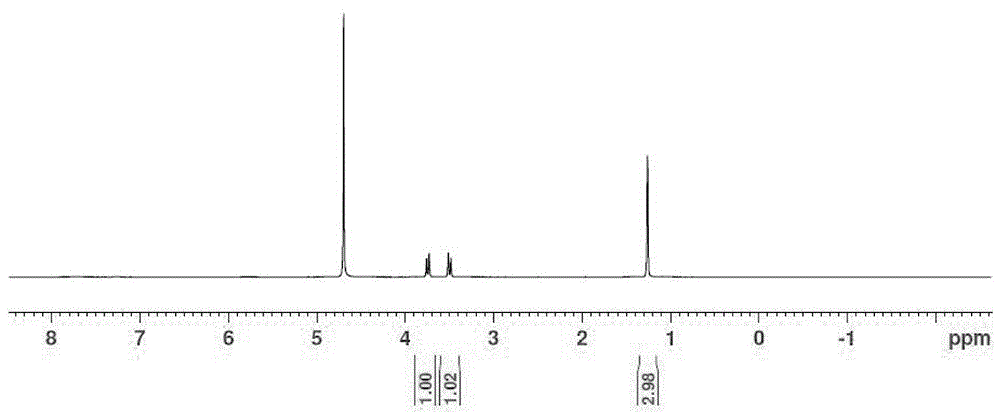
图1为本发明产品2-甲基-l-丝氨酸氢谱。
图2为本发明中间体1氢谱。
图3为本发明中间体3氢谱。
图4为本发明中间体4氢谱。
图5为本发明产品2-甲基-d-丝氨酸氢谱。
具体实施方式
下面结合具体实施例对本发明进一步说明,这些实例不应解释为对本发明的限制。
实施例1
(1):将cbz-d-丙氨酸200g溶于1l四氢呋喃中,加入157g苯甲醛二甲缩醛,通入氮气置换体系,冰盐浴降温至-15℃,滴入106.7g氯化亚砜,控制温度低于0℃,滴完搅拌至温度至-15℃,分批次加入122g无水氯化锌,控制温度低于0℃,加完搅拌4小时,加入1l水淬灭反应,然后用600ml乙酸乙酯萃取,有机相依次用水和饱和碳酸氢钠溶液洗涤至中性,无水硫酸钠干燥,浓缩后用300ml异丙醚和200ml石油醚重结晶,过滤,干燥得到129g中间体1,氢谱见图2,白色固体,收率46%。1hnmr(400mhz,cdcl3):δ7.50~7.10(m,10h),6.64(s,1h),5.14(s,2h),4.49(m,1h),1.58(m,3h)。
(2):将100g中间体1溶于500ml四氢呋喃中,通入氮气置换体系,干冰丙酮浴降温至-78℃后,滴入485ml六甲基二硅基胺基锂(1mol/l),控制温度-65~-50℃,约2小时滴完,搅拌1.5小时,滴入61g氯甲基苄醚,滴完搅拌半小时,然后自然升温至20~25℃,继续搅拌1小时,检测中间体1反应完全,加600ml冰水淬灭反应,再加入600ml乙酸乙酯搅拌后分层。有机相再用300ml水和饱和食盐水洗涤一次,干燥,浓缩,得到166g粗品中间体2,为淡黄色油状物,直接下一步。
(3):将166g粗品中间体2溶于330ml四氢呋喃,加入1l5.4%质量百分浓度的氢氧化锂水溶液,室温搅拌反应12小时,检测中间体2反应完全,反应液用300ml乙酸乙酯洗涤二次后,用6mol/l盐酸调ph至3~4,加入900ml乙酸乙酯萃取水相,乙酸乙酯相再用500ml水洗涤一次,干燥,浓缩得到74.3g中间体3,氢谱见图3,为棕色油状物,二步收率67%,手性检测合格ee>99%。1hnmr(400mhz,cdcl3):δ7.41~7.18(m,10h),5.79(s,1h),5.09(s,2h),4.52(s,2h),3.79(s,2h),1.58(s,3h)。
(4):将74.3g中间体3,溶于300ml甲醇与80ml水的混合体系中,加入7.5g钯炭(钯含重量10%),通入氢气,升温至60℃,压力为3mpa,反应5~6小时,检测反应完全,冷却至室温,过滤,固体用250ml水洗涤,滤液和洗液合并浓缩后得到粗品。粗品加入200ml丙酮打浆,过滤,烘干,得到最终产物2-甲基-l-丝氨酸20.7g,氢谱见图1,为白色固体,收率80%。旋光+4.5(c=1,h2o),1hnmr(400mhz,d2o):δ3.74(m,h),3.50(m,1h),1.26(s,3h)。
实施例2:
(1):将cbz-d-丙氨酸71kg溶于350l四氢呋喃中,加入55.7kg苯甲醛二甲缩醛,通入氮气置换体系,冰盐浴降温至-15℃,滴入37.9kg氯化亚砜,控制温度低于0℃,滴完搅拌至温度冷却至-15℃,分批次加入43.3kg无水氯化锌,控制温度低于0℃,加完搅拌6小时。加入300l冰水淬灭反应,然后用200l乙酸乙酯萃取,有机相依次用300l水洗一次,约500l饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥,旋干,加入90l异丙醚和60l石油醚重结晶,过滤,35℃左右烘干得到36.3kg中间体1,氢谱见图2,为白色固体,收率36%。
(2):将31kg中间体1溶于150l四氢呋喃中,通入氮气置换体系,液氮降温至-78℃,滴入150l六甲基二硅基胺基锂(1mol/l),控制温度-65~-50℃,约2小时滴完,搅拌1.5小时,控制温度在-75~-70℃,滴入19kg氯甲基苄醚,滴完搅拌半小时,然后自然升温至20~25℃,大约5小时,继续搅拌1小时,加入200l冰水淬灭反应,再加入200l乙酸乙酯萃取,分层,有机相再用100l水洗涤一次,干燥,浓缩,得到47.5kg粗品中间体2,为淡黄色油状物,直接下一步。
(3):将47.5kg粗品中间体2溶于80l四氢呋喃,加入预先溶解好的240l氢氧化锂(17.5kg)溶液,加完室温搅拌12小时,检测中间体2反应完全,用乙酸乙酯洗涤三次(3*100l),水层用6mol/l盐酸(约90l)调ph至3~4左右,加入200l乙酸乙酯萃取水相,乙酸乙酯相再用200l水洗涤一次,干燥,浓缩得到21.3kg中间体3,氢谱见图3,为棕色油状物,二步收率62%。
(4):将650g中间体3,溶于2.2l甲醇与650ml水的混合体系中,加入70g钯炭(钯含重量10%),通入氢气,升温至60℃,压力为3mpa,反应5~6小时,检测反应完全,冷却至室温,过滤,固体用2l水洗涤,滤液和洗液合并浓缩后得到粗品。粗品加入200ml丙酮打浆,过滤,烘干,得到最终产物2-甲基-l-丝氨酸194g,氢谱见图1,为白色固体,收率86%,手性检测合格ee>99%。
实施例3:步骤(2)所用碱为六甲基二硅基胺基钠,其余同实施例1,步骤(2)和(3)合并收率35%。
实施例4:步骤(2)所用碱为六甲基二硅基胺基钾,其余同实施例1.步骤(2)和(3)合并收率30%。
实施例5:步骤(2)所用碱为六甲基二硅基胺基钠,其余同实施例1,步骤(2)和(3)合并收率35%。
实施例6:步骤(3)所用碱为氢氧化钠,其余同实施例1,步骤(2)和(3)合并收率60%。
实施例7:步骤(3)所用碱为氢氧化钾,其余同实施例1,步骤(2)和(3)合并收率55%。
实施例8:步骤(4)反应温度为室温,压力为1大气压,反应时间延长到20小时,其余同实施例1,收率80%。
实施例9:
(1):将cbz-l-丙氨酸400g溶于2l四氢呋喃中,加入314g苯甲醛二甲缩醛,通入氮气置换体系,冰盐浴降温至-15℃,滴入213.4g氯化亚砜,控制温度低于0℃,滴完搅拌至温度至-15℃,分批次加入244g无水氯化锌,控制温度低于0℃,加完搅拌4小时,加入2l水淬灭反应,然后用1.2l乙酸乙酯萃取,有机相依次用水和饱和碳酸氢钠溶液洗涤至中性,无水硫酸钠干燥,浓缩后用600ml异丙醚和400ml石油醚重结晶,过滤,干燥得到257g中间体4,氢谱见图4,白色固体,收率46%。1hnmr(400mhz,cdcl3):δ7.50~7.10(m,10h),6.64(s,1h),5.14(s,2h),4.49(m,1h),1.58(m,3h)。
(2):将100g中间体4溶于500ml四氢呋喃中,通入氮气置换体系,干冰丙酮浴降温至-78℃后,滴入485ml六甲基二硅基胺基锂(1mol/l),控制温度低于-65~-50℃,约2小时滴完,搅拌1.5小时,滴入61g氯甲基苄醚,滴完搅拌半小时,然后自然升温至20~25℃,继续搅拌1小时,检测中间体1反应完全,加600ml冰水淬灭反应,再加入600ml乙酸乙酯搅拌后分层。有机相再用300ml水和饱和食盐水洗涤一次,干燥,浓缩,得到170g粗品中间体5,为淡黄色油状物,直接下一步。
(3):将170g粗品中间体5溶于330ml四氢呋喃,加入1l5.4%质量百分浓度的氢氧化锂水溶液,室温搅拌反应12小时,检测中间体2反应完全,反应液用300ml乙酸乙酯洗涤二次后,用6mol/l盐酸调ph至3~4,加入900ml乙酸乙酯萃取水相,乙酸乙酯相再用500ml水洗涤一次,干燥,旋干得到77g中间体6,为棕色油状物,二步收率68%,手性检测合格ee>99%。1hnmr(400mhz,cdcl3):δ7.41~7.20(m,10h),5.80(s,1h),5.10(s,2h),4.53(s,2h),3.80(s,2h),1.59(s,3h)。
(4):将77g中间体6,溶于300ml甲醇与80ml水的混合体系中,加入8.0g钯炭(钯含重量10%),通入氢气,升温至60℃,压力为3mpa,反应5~6小时,检测反应完全,冷却至室温,过滤,固体用250ml水洗涤,滤液和洗液合并浓缩后得到粗品。粗品加入200ml丙酮打浆,过滤,烘干,得到最终产物2-甲基-d-丝氨酸22.3g,氢谱见图5,为白色固体,收率83%。旋光-5.1(c=1,h2o),1hnmr(400mhz,d2o):δ3.77(m,h),3.52(m,1h),1.28(s,3h)。
- 还没有人留言评论。精彩留言会获得点赞!