一种具有微相分离结构的含偶氮苯基侧链型液晶聚合物及其制备方法与流程
本发明属于液晶功能化聚合物合成与应用领域,涉及一种含偶氮苯基侧链液晶聚合物及其制备方法,尤其涉及一种具有微相分离结构的含偶氮苯基侧链型液晶聚合物及其制备方法。
背景技术:
随着纳米技术的迅速发展,开发具有精细结构的纳米材料是近年来的研究热点之一。分子自组装是液晶高分子精细结构调控的典型的方式。合成具有不相容性的聚合物是自组装的简单手段之一,组分的变化可呈现不同的相分离结构。因此,微观相分离具有许多独特的结构和应用前景,受到人们广泛的关注。液晶高分子的微相分离通常发生在嵌段共聚物中。通常具有刚柔结构组成的嵌段共聚物由于热力学性质的差异容易产生微观相分离,研究表明,微观相分离也可能存在于均聚物中两不相容性的部分。较少有涉及到基于均聚物主链接枝的液晶聚合物的微相分离的报道。
智能材料作为一种智能仿生材料,能够对外部环境刺激(如光、电场、磁场、压力、温度等)做出响应,将相应的能量转化为机械能,使自身的形状能发生明显变化的新型材料,柔性执行器件的构筑在软体机器人、3d打印、人造肌肉等领域得到了广泛关注。侧链液晶聚合物(sclcps)具有较好的分子结构可设计性,在性质上既具有高分子特性,又结合了液晶的优良特性赋予材料分子间的协同作用以及各向异性,因而广泛用于智能材料的开发设计。
光是一种清洁能源,来源丰富,可以通过远程非接触方式精确快速地进行调控,还可以通过改变波长、强度、偏振方向等参数进行调控,因而倍受科研人员的青睐。偶氮苯及其衍生物由于具有独特的光致可逆异构化特性和优越的光诱导取向能力,因而成为目前使用最广泛的光响应性基团。此外偶氮苯基团是一个长径比很大的刚性棒状结构,非常适合用作液晶基元,这使得偶氮苯化合物通过合理的结构设计可以表现出液晶相。含偶氮苯基侧链液晶聚合物结合液晶的排列有序及偶氮苯基的光致异构的优良特性。此外,偶氮苯及其衍生物在一定波长的照射下,其可逆的顺反异构化可引起液晶基元排列的变化,可有效触发液晶高分子发生宏观形变,是一种性能优良的智能材料,在工业、医疗等领域有着广泛的应用,用于柔性执行器、光信息存储材料,实现信息的存储与擦除,及微型反应器的构筑和开发。
智能材料在结构上一般是由柔软及其易于变形的材料组成,因而赋予其较好的加工性能,并且材料的形变具有良好的可逆性和可重复性。但在实际应用中由于材料自身的柔软性深受机械强度的影响,因此要求材料具有一定的刚性。
液晶高分子材料所展现的优异性能与其结构的特殊性密不可分。通过对聚合物主链、间隔基、液晶基元、聚合物的多分散指数(pdi)等因素的协同设计可实现sclcps性能的精确调控。首先,对于sclcps结构-性能‘构-效’关系的研究,可控窄分布的sclcps的合成是必要的。
聚苯乙烯(ps)作为一种刚性主链,具有高强度、高模量及较好的光学性能,能够提供较好的机械稳定性、加工性能和透明度。此外,ps可进行可控的活性阴离子聚合,定制合成不同分子量窄分布的模板主链。聚苯乙烯本身无官能团反应位点,因此无法进行功能化反应。含硅氢功能化的聚苯乙烯可通过活性阴离子方法定制合成,结合高效的硅氢加成反应接枝不同结构的液晶单体。此外,液晶的有序结构可导致液晶聚合物组分的不相容,有望构筑不同的相分离结构。
本发明通过系列反应步骤合成含偶氮苯基的液晶单体m,并且通过活性阴离子聚合制备基于聚苯乙烯(ps)的含硅氢功能化的模板主链(pvpdms),然后结合高效的硅氢加成反应制备含偶氮苯基侧链液晶聚合物pvpdms-m,研究该液晶聚合物的光响应性能及液晶性能。
技术实现要素:
针对现有技术存在的问题,本发明将含偶氮苯的液晶单体m,引入到si-h功能化的聚合主链pvpdms中,构筑一种具有微相分离结构的含偶氮苯基侧链型液晶聚合物。并分析该液晶聚合物的光响应性能及液晶性能。
为实现上述目的,本发明采用如下技术方案:
一种具有微相分离结构的含偶氮苯基侧链型液晶聚合物pvpdms-m,是由主链基于苯乙烯的含硅氢功能化的聚合物主链pvpdms与一种末端双键的含偶氮苯基的液晶单体m通过硅氢加成反应构筑而成。pvpdms-m的数均分子量(mn)为11.8-32.6kg·mol-1,分子量分布指数(pdi)为1.30-1.42,液晶形成区间为[tg57-77℃-lc-ti186-218℃],所合成的液晶为具有双分子层微观结构的近晶a相,pvpdms-m在结构上包含苯乙烯硬段、烷烃链软段和液晶核硬段,液晶聚合物具有微相分离结构。液晶单体m和液晶聚合物pvpdms-m由于结构中含有偶氮苯基团其形成的液晶具有光响应性,pvpdms-m形成的液晶在温度区间57℃-218℃内具有光响应性。
所述的pvpdms-m结构式如下:
所述的含偶氮苯基的液晶单体m的液晶形成区间为[tm113℃-lc-ti218℃],液晶单体m为近晶a相转变为向列相;所述的液晶单体m形成的液晶在温度区间115℃-218℃内也具有光响应性。所述的液晶单体m其结构式如下:
所述的硅氢(si-h)功能化的聚合物主链pvpdms,由si-h官能化聚合单体vpdms通过活性阴离子聚合制得。
一种具有微相分离结构的含偶氮苯基侧链型液晶聚合物pvpdms-m的制备方法,包括以下步骤:
第一步,合成液晶单体m
1.1)合成对羟基偶氮苯甲酸乙酯azo-oh
将对氨基甲酸乙酯加入到稀释的hcl水溶液中形成氨基盐酸盐,将其置于0℃的冰浴中,将亚硝酸钠水溶液滴加至上述的盐酸盐水溶液中形成重氮盐。滴加完后,搅拌反应30-60min,溶液澄清。将苯酚溶解至naoh溶液中,置于0℃的冰浴中。将上述的重氮盐水溶液缓慢滴加至苯酚的naoh溶液中,滴加完后,继续反应60-120min。反应结束后,反应物用5%的盐酸酸化,酸化至ph=5,产生大量的黄色固体,抽滤,将得到的固体用乙醇重结晶,得到黄色粉末azo-oh。
1.2)合成中间产物a
将对羟基苯甲酸溶解在ch2cl2和吡啶的混合溶剂中得到混合体系,将10-十一烯酰氯于室温缓慢滴加至上述的混合体系中。滴加完后,回流反应6-12h。反应结束后减压旋蒸除去溶剂,将剩余物倒入大量稀释的hcl水溶液中静置过夜。抽滤得到粗产物,用大量温水清洗粉末,烘干后,热乙醇重结晶,得到白色片状晶体a。
1.3)合成中间产物b
将得到的中间产物a溶解至socl2中,60-80℃加热反应6-10h。反应完毕后,通过减压蒸馏装置脱除过量的socl2,得到淡黄色液体b。
1.4)合成液晶单体m
将1.1)合成得到的中间产物azo-oh溶解在ch2cl2和吡啶的混合溶剂中得到混合体系,将中间产物b于室温缓慢滴加至上述的混合体系中。滴加完后,回流反应6-12h。反应结束后减压旋蒸除去溶剂,将剩余物倒入大量稀释的hcl水溶液中静置过夜。抽滤得到粗产物,用大量温水清洗粉末,烘干后,热乙醇重结晶,得到黄色粉末m。
所述的液晶单体m的总的合成路线如下:
第二步,合成聚合单体vpdms
在无水无氧的氩气保护下,将镁屑加入到干燥的thf中,先滴加少量的4-氯苯乙烯,使用碘粒引发格式反应。置于0℃的冰浴中,继续滴加二甲基氯硅烷。滴加完成后,温度升高至65℃回流反应90min。反应完成后,冷却至室温。将反应液导入到另一个无水无氧的氩气保护的反应烧瓶中,然后将二甲基氯硅烷缓慢滴加至上述的反应液中,滴加完后室温反应12h。反应完毕后,将混合物用乙醚萃取,收集有机层,水洗3-4次,然后用无水硫酸镁干燥抽滤,将得到的滤液旋蒸除去溶剂thf和乙醚,剩余物用柱层析的方法进行分离纯化。将纯化得到的产物用氢化钙搅拌干燥5h后减压蒸馏,将减压蒸馏的产物加入到二丁基镁的庚烷溶液中搅拌3h后减压蒸馏,得到纯净的聚合单体vpdms。
所述的聚合单体vpdms,合成路线为:
第三步,合成si-h功能化聚合主链pvpdms
在无水无氧、氩气保护的手套箱中,采用活性阴离子聚合的方法,以苯为溶剂,以sec-buli作为引发剂,引发vpdms单体的聚合反应。其中,溶剂和单体的体积比为10-20:1。根据vpdms单体的投料质量和设计的聚合物分子量加入定量的sec-buli引发剂,具体为:
在聚合瓶中顺序加入溶剂苯,sec-buli和vpdms单体,反应溶液迅速变成橙黄色。25℃下反应5h,最后加入精制的异丙醇终止反应。将反应溶液沉于大量的甲醇中,抽滤,得到的固体粉末干燥至恒重,得到不同分子量的si-h功能化聚合主链pvpdms。所述的引发剂的浓度为0.3-0.5mol/l。
第四步,合成含偶氮苯基侧链型液晶聚合物
4.1)在无水无氧、氩气保护下的手套箱中,将第三步制备得到的聚合主链pvpdms的sih与第一步制备得到的液晶单体m以摩尔比为1:1-1:2置于安瓿瓶中,加入无水无氧处理后的甲苯,搅拌均匀后,滴加3-6滴卡斯特催化剂,引发硅氢加成反应。
所述的卡斯特催化剂溶解于二甲苯溶液中,质量浓度为2%。
所述的每200mgsih聚合物主链pvpdms对应加入20-50ml甲苯。
4.2)从手套箱中取出,45-60℃油浴中反应48-72h。反应结束后,减压旋蒸脱除溶剂,将反应物用少量的四氢呋喃溶解,向溶液中滴加乙醚有固体析出,抽滤,将得到的固体重复上述操作2-3次,得到纯净的侧链型液晶聚合物pvpdms-m。
液晶单体和液晶聚合物的光响应性能分析:配置液晶单体和液晶聚合物的四氢呋喃稀溶液用紫外点光源(6w)在溶液的上方垂直照射不同的时间,进行紫外光谱检测。
液晶性能分析:液晶织构通过配备有热台和数字相机的偏光显微镜(pom)观察。以凝胶渗透色谱仪(gpc)测定聚合物的分子量和分子量分布指数(重均分子量与数均分子量之比),采用示差量热扫描仪(dsc)测定液晶单体的熔点tm和清亮点ti,以及聚合物的玻璃化转变温度tg和清亮点ti。
本发明的有益效果为:本发明设计合成液晶单体m,将其引入到基于ps的硅氢功能化的聚合物主链pvpdms中,构筑一种含偶氮苯基侧链液晶聚合物。液晶单体和液晶聚合物膜都具有光响应性,液晶织构也表现出光响应性。液晶单体m的液晶形成温度范围(δt=ti-tm)为105℃,在液晶形成区间,分别形成近晶a相以及向列相液晶典型的焦锥织构和纹影织构。pvpdms-m:tg1为21-25℃,tg2为57-77℃,ti为186-218℃,δt(ti-tg2)为123-156℃,在液晶形成区间,形成了近晶a相。该液晶聚合物在结构上具有“软段-硬段”类似于sbs热塑性弹性体产生微相分离结构。
附图说明
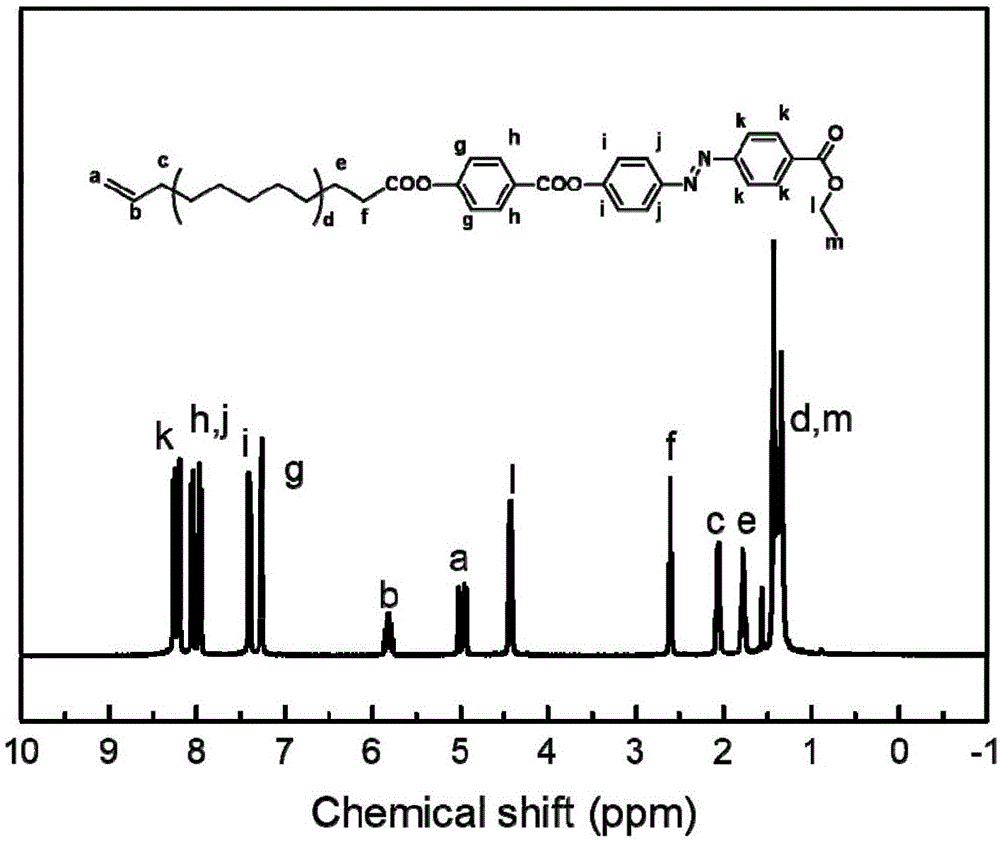
图1(a)为液晶单体m的1hnmr谱图及峰位归属。
图1(b)为液晶单体m、主链pvpdms与代表性的液晶聚合物pvpdms3.5k-m的1hnmr谱图对比及峰位归属。
图2为液晶单体m、pvpdms3.5k-m在thf溶液中的紫外光谱图。其中,(a)为液晶单体m,(b)为pvpdms3.5k-m。
图3为液晶单体m的偏光织构图片:(a)155℃(b)170℃,(c)180℃,(d)190℃,(e)202℃,(f)210℃,其中,图中上方从左到右依次为(a)、(b)、(c),图中下方从左到右依次为(d)、(e)、(f)。
图4为液晶聚合物pvpdms-m的偏光织构图片,(a)升温160℃,(b)为172℃。
图5为液晶聚合物pvpdms-m温度接近ti的不同放大倍数的偏光照片
图6为液晶单体m、液晶聚合物pvpdms3.5k-m的液晶织构在紫外光照前后的偏光照片。
图7为液晶单体m的dsc曲线。
图8为液晶聚合物pvpdms-m的dsc曲线(二次升温)。
具体实施方式
以下结合具体实施例对本发明做进一步说明。
实施例1
第一步,合成液晶单体m
1.1)合成对羟基偶氮苯甲酸乙酯azo-oh
在500ml的烧杯中,加入对氨基甲酸乙酯(10g,0.0605mol),去离子水30ml和浓盐酸30ml(稀释至120ml),搅拌至混合均匀,将其置于0℃的冰浴中。将亚硝酸钠(5.15g,0.075mol)溶解至30ml的去离子水中,滴加至上述的浓盐酸和对氨基苯甲酸乙酯的混合体系中,体系逐渐澄清,形成黄色溶液。滴加完后,继续搅拌反应30min。在500ml烧杯中加入苯酚(6.84g,0.073mol),naoh(7.2g,0.18mol)和去离子水20ml,置于0℃的冰浴中,慢慢滴加上述的黄色溶液,溶液中产生黄色固体,滴加完毕后,继续搅拌反应60min。反应结束后,反应物用5%的盐酸酸化,酸化至ph=5,产生大量的黄色固体。抽滤,用蒸馏水洗涤2-3次,将得到的固体用乙醇重结晶,得到黄色粉末azo-oh。
1.2)合成中间产物a
将对羟基苯甲酸(8.6g,0.06mol)溶解在40ml的二氯甲烷和4ml吡啶的混合溶剂中,将10-十一烯酰氯(10.1g,0.05mol)于室温缓慢滴加至上述的混合体系中。滴加完后,回流反应6h。反应结束后减压旋蒸除去溶剂,将得到的混合物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粗产物2-3次,烘干,热乙醇重结晶,得到白色片状晶体a。
1.3)合成中间产物b
将中间产物a(5g,0.016mol)溶解至socl2(5.7g,0.048mol)中,60℃加热反应10h。反应完毕后,通过减压蒸馏装置脱除过量的socl2,得到淡黄色液体b。
1.4)合成液晶单体m
将azo-oh(4.3g,0.016mol)溶解在50ml的ch2cl2和3ml吡啶的混合溶剂中,将中间产物b(5.17g,0.016mol)于室温缓慢滴加至上述的混合体系中。滴加完后,加热回流反应6h。反应结束后减压旋蒸除去溶剂,将剩余物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粉末2-3次,烘干后,热乙醇重结晶,得到黄色粉末m。
第二步,合成聚合单体vpdms
合成聚合单体vpdms
在无水无氧的氩气保护下,将镁屑(52.6g,2.16mol)加入到干燥的1000ml的四氢呋喃中。在恒压滴液漏斗中加入4-氯苯乙烯(100g,0.72mol),先滴加少量的4-氯苯乙烯,加入碘粒引发格式反应。置于0℃的冰浴中,继续滴加二甲基氯硅烷。滴加完毕后,温度升高至65℃回流反应90min。反应完毕后,将反应冷却至室温。将反应液导入到另一个无水无氧的氩气保护的三口烧瓶中,然后将二甲基氯硅烷(67.7g,0.72mol)缓慢滴加至上述的反应液中,滴加完后室温反应12h。反应完毕后,将混合物用乙醚萃取,收集有机层,水洗3-4次,然后用无水硫酸镁干燥抽滤,将得到的滤液旋蒸除去溶剂thf和乙醚,剩余物用柱层析的方法进行分离纯化,以正己烷为展开剂。将过柱纯化得到的产物用氢化钙搅拌干燥5h后减压蒸馏,将减压蒸馏的产物加入到二丁基镁的庚烷溶液中搅拌3h后减压蒸馏,得到纯净的聚合单体vpdms。
第三步,合成si-h功能化聚合物主链pvpdms
在无水无氧、氩气保护的手套箱中,采用活性阴离子聚合的方法,以苯为溶剂,以sec-buli作为引发剂,引发单体vpdms的聚合反应。在聚合瓶中依次加入20ml苯,引发剂sec-buli(0.3483mol·l-1,0.36ml和0.62ml)和vpdms单体(1g,0.0062mol),反应溶液迅速变成橙黄色。25℃下反应5h,加入精制的异丙醇终止反应。将反应溶液沉胶于400ml的甲醇中,抽滤,得到的固体粉末干燥至恒重,得到si-h功能化聚合主链pvpdms。聚合物数均分子量为4.6kg·mol-1和8.3kg·mol-1,分子量分布指数分别为1.11和1.09。
第四步,合成含偶氮苯基侧链型液晶聚合物
合成侧链液晶聚合物pvpdms-m
在无水无氧、氩气保护下的手套箱中,将pvpdms4.6k和pvpdms8.3k(200mg,1.21mmolsi-h)加入到100ml安瓿瓶中。称取液晶单体m(0.58g,1.33mmolc=c)溶解至30ml无水无氧处理后的甲苯中,混合均匀后,加入到上述的安瓿瓶中,滴加4滴卡斯特催化剂(所用的卡斯特催化剂溶解于二甲苯溶液中,质量浓度为2%),引发硅氢加成反应。从手套箱中取出,45℃油浴中反应72h。反应结束后,减压旋蒸脱除溶剂,将反应物用少量的四氢呋喃溶解,向溶液中滴加乙醚有固体析出,抽滤,将得到的固体重复上述操作2-3次,得到纯净的侧链型液晶聚合物pvpdms4.6k-m和pvpdms8.3k-m。液晶聚合物数均分子量为19.2kg·mol-1和32.6kg·mol-1,分子量分布指数为1.30和1.39。液晶聚合物产生了微相分离,都具有两个玻璃化转变温度,分别为聚合主链产生的玻璃化转变温度tg1为21℃和25℃,侧链液晶导致的tg2为60℃和62℃。
配置液晶单体和液晶聚合物的四氢呋喃稀溶液用紫外点光源(6w)在溶液的上方垂直照射不同的时间,对样品在紫外光照前后进行紫外光谱检测。
液晶单体及液晶聚合物的液晶织构通过带热台和数字相机的偏光显微镜观察,升温或降温速率为2℃/min。。
分别称取5-10mg的液晶单体m和pvpdms-m进行dsc曲线的测定,样品的测定在氮气环境下进行,升温和降温速度均为20℃·min-1;选取pvpdms-m的二次升温曲线记录玻璃化转变温度(tg1和tg2)和液晶相各向同性转变温度(ti),液晶相温度范围δt=ti-tg2;选取液晶单体一次升温与一次降温曲线中的熔点温度(tm)、清亮点温度(ti),δt=ti-tm。
图1(a)为液晶单体m的1hnmr谱图及峰归属,图中:
m:1hnmr(400mhz,cdcl3)δ8.26(dd,j=20.4,8.4hz,4h,ar-h),8.07(d,j=8.6hz,2h,ar-h),7.98(d,j=8.3hz,2h,ar-h),7.43(d,j=8.6hz,2h,ar-h),7.34-7.28(m,2h,ar-h),5.84(dd,j=17.0,10.3hz,ch2=ch-),5.00(dd,j=24.3,13.7hz,2h,ch2=ch-),4.45(q,j=7.1hz,-cooch2ch3-),2.63(t,j=7.5hz,2h,-ch2coo-),2.08(d,j=6.9hz,2h,ch2=ch-ch2-),1.89-1.73(m,2h,-ch2ch2coo-),1.43(dd,j=26.4,19.2hz,13h,10hinalkanechainand-cooch2ch3).
图1(b)为液晶单体m、主链pvpdms与液晶聚合物pvpdms-m的1hnmr谱图对比,表明含偶氮苯基团的液晶单体m成功接枝到了pvpdms聚合物主链中,成功合成了含偶氮苯基侧链型液晶聚合物pvpdms-m。由图2可知:液晶单体m和液晶聚合物pvpdms-m在紫外光照下偶氮苯基团能够发生结构的顺反异构,具有光响应性。图3可知:液晶单体m在液晶形成区间呈现了明显的焦锥织构和纹影织构。由图4、图5可知:侧链型液晶聚合物pvpdms-m具有明显的液晶织构,具有液晶性。由图6可知:液晶单体和液晶聚合物形成的液晶具有光响应性。图7可知:液晶单体m呈现了由近晶a相到向列相的转变tsn。图8可知pvpdms-m在dsc曲线中呈现两个明显的玻璃化转变温度tg1和tg2及清亮点温度ti。由图5和图8可知:pvpdms-m具有微相分离结构。
实施例2:
第一步,合成液晶单体m
1.1)合成对羟基偶氮苯甲酸乙酯azo-oh
在500ml的烧杯中,加入对氨基甲酸乙酯(15g,0.0908mol),去离子水45ml和浓盐酸45ml(稀释至150ml),搅拌至混合均匀,将其置于0℃的冰浴中。将亚硝酸钠(7.7g,0.113mol)溶解至45ml的去离子水中,滴加至上述的浓盐酸和对氨基苯甲酸乙酯的混合体系中,体系逐渐澄清,形成黄色溶液。滴加完后,继续搅拌反应45min。在500ml烧杯中加入苯酚(10.3g,0.110mol),naoh(10.8g,0.27mol)和去离子水30ml,置于0℃的冰浴中,慢慢滴加上述的黄色溶液,溶液中产生黄色固体,滴加完毕后,继续搅拌反应90min。反应结束后,反应物用5%的盐酸酸化,酸化至ph=5,产生大量的黄色固体。抽滤,用蒸馏水洗涤2-3次,将得到的固体用乙醇重结晶,得到黄色粉末azo-oh。
1.2)合成中间产物a
将对羟基苯甲酸(17.3g,0.13mol)溶解在75ml的二氯甲烷和8ml吡啶的混合溶剂中,将10-十一烯酰氯(20.2g,0.1mol)于室温缓慢滴加至上述的混合体系中。滴加完后,回流反应9h。反应结束后减压旋蒸除去溶剂,将得到的混合物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粗产物2-3次,烘干,热乙醇重结晶,得到白色片状晶体a。
1.3)合成中间产物b
将中间产物a(5g,0.016mol)溶解至socl2(5.7g,0.048mol)中,70℃加热反应8h。反应完毕后,通过减压蒸馏装置脱除过量的socl2,得到淡黄色液体b。
1.4)合成液晶单体m
将azo-oh(6.5g,0.024mol)溶解在75ml的ch2cl2和5ml吡啶的混合溶剂中,将中间产物b(7.8g,0.024mol)于室温缓慢滴加至上述的混合体系中。滴加完后,加热回流反应9h。反应结束后减压旋蒸除去溶剂,将剩余物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粉末2-3次,烘干后,热乙醇重结晶,得到黄色粉末m。
第二步,合成聚合单体vpdms
在无水无氧的氩气保护下,将镁屑(52.6g,2.16mol)加入到干燥的1000ml的四氢呋喃中。在恒压滴液漏斗中加入4-氯苯乙烯(100g,0.72mol),先滴加少量的4-氯苯乙烯,加入碘粒引发格式反应。置于0℃的冰浴中,继续滴加二甲基氯硅烷。滴加完毕后,温度升高至65℃回流反应90min。反应完毕后,将反应冷却至室温。将反应液导入到另一个无水无氧的氩气保护的三口烧瓶中,然后将二甲基氯硅烷(67.7g,0.72mol)缓慢滴加至上述的反应液中,滴加完后室温反应12h。反应完毕后,将混合物用乙醚萃取,收集有机层,水洗3-4次,然后用无水硫酸镁干燥抽滤,将得到的滤液旋蒸除去溶剂thf和乙醚,剩余物用柱层析的方法进行分离纯化,以正己烷为展开剂。将过柱纯化得到的产物用氢化钙搅拌干燥5h后减压蒸馏,将减压蒸馏的产物加入到二丁基镁的庚烷溶液中搅拌3h后减压蒸馏,得到纯净的聚合单体vpdms。
第三步,合成si-h功能化聚合物主链pvpdms
在无水无氧、氩气保护的手套箱中,采用活性阴离子聚合的方法,以苯为溶剂,以sec-buli作为引发剂,引发单体vpdms的聚合反应。在聚合瓶中依次加入20ml苯,引发剂sec-buli(0.3845mol·l-1,0.59ml)和vpdms单体(0.5g,0.0031mol),反应溶液迅速变成橙黄色。25℃下反应5h,加入精制的异丙醇终止反应。将反应溶液沉胶于400ml的甲醇中,抽滤,得到的固体粉末干燥至恒重,得到si-h功能化聚合主链pvpdms。聚合物数均分子量为2.2kg·mol-1,分子量分布指数为1.17。
第四步,合成含偶氮苯基侧链型液晶聚合物
在无水无氧、氩气保护下的手套箱中,将pvpdms2.2k(200mg,1.21mmolsi-h)加入到100ml安瓿瓶中。称取液晶单体m(1.0g,2.42mmolc=c)溶解至50ml无水无氧处理后的甲苯中,混合均匀后,加入到上述的安瓿瓶中,滴加4滴卡斯特催化剂(所用的卡斯特催化剂溶解于二甲苯溶液中,质量浓度为2%),引发硅氢加成反应。从手套箱中取出,50℃油浴中反应60h。反应结束后,减压旋蒸脱除溶剂,将反应物用少量的四氢呋喃溶解,向溶液中滴加乙醚有固体析出,抽滤,将得到的固体重复上述操作2-3次,得到纯净的侧链型液晶聚合物pvpdms-m。液晶聚合物数均分子量为11.8kg·mol-1,分子量分布指数为1.42。该液晶聚合物产生了微相分离,具有两个玻璃化转变温度,分别为聚合主链产生的玻璃化转变温度tg1为24℃,侧链液晶导致的tg2为57℃。
实施例3:
第一步,合成液晶单体m
1.1)合成对羟基偶氮苯甲酸乙酯azo-oh
在500ml的烧杯中,加入对氨基甲酸乙酯(20g,0.121mol),去离子水60ml和浓盐酸60ml(稀释至200ml),搅拌至混合均匀,将其置于0℃的冰浴中。将亚硝酸钠(11.6g,0.170mol)溶解至60ml的去离子水中,滴加至上述的浓盐酸和对氨基苯甲酸乙酯的混合体系中,体系逐渐澄清,形成黄色溶液。滴加完后,继续搅拌反应60min。在500ml烧杯中加入苯酚(15.5g,0.165mol),naoh(16.2g,0.405mol)和去离子水45ml,置于0℃的冰浴中,慢慢滴加上述的黄色溶液,溶液中产生黄色固体,滴加完毕后,继续搅拌反应120min。反应结束后,反应物用5%的盐酸酸化,酸化至ph=5,产生大量的黄色固体。抽滤,用蒸馏水洗涤2-3次,将得到的固体用乙醇重结晶,得到黄色粉末azo-oh。
1.2)合成中间产物a
将对羟基苯甲酸(34.5g,0.25mol)溶解在150ml的二氯甲烷和16ml吡啶的混合溶剂中,将10-十一烯酰氯(40.5g,0.2mol)于室温缓慢滴加至上述的混合体系中。滴加完后,加热回流反应12h。反应结束后减压旋蒸除去溶剂,将得到的混合物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粗产物2-3次,烘干,热乙醇重结晶,得到白色片状晶体a。
1.3)合成中间产物b
将中间产物a(5g,0.016mol)溶解至socl2(5.7g,0.048mol)中,80℃加热反应6h。反应完毕后,通过减压蒸馏装置脱除过量的socl2,得到淡黄色液体b。
1.4)合成液晶单体m
将azo-oh(8.6g,0.032mol)溶解在100ml的ch2cl2和6ml吡啶的混合溶剂中,将中间产物b(10.3g,0.032mol)于室温缓慢滴加至上述的混合体系中。滴加完后,加热回流反应12h。反应结束后减压旋蒸除去溶剂,将剩余物倒入大量稀释的hcl水溶液中酸化过夜。抽滤得到粗产物,用大量50℃温水清洗粉末2-3次,烘干后,热乙醇重结晶,得到黄色粉末m。
第二步,合成聚合单体vpdms
在无水无氧的氩气保护下,将镁屑(52.6g,2.16mol)加入到干燥的1000ml的四氢呋喃中。在恒压滴液漏斗中加入4-氯苯乙烯(100g,0.72mol),先滴加少量的4-氯苯乙烯,加入碘粒引发格式反应。置于0℃的冰浴中,继续滴加二甲基氯硅烷。滴加完毕后,温度升高至65℃回流反应90min。反应完毕后,将反应冷却至室温。将反应液导入到另一个无水无氧的氩气保护的三口烧瓶中,然后将二甲基氯硅烷(67.7g,0.72mol)缓慢滴加至上述的反应液中,滴加完后室温反应12h。反应完毕后,将混合物用乙醚萃取,收集有机层,水洗3-4次,然后用无水硫酸镁干燥抽滤,将得到的滤液旋蒸除去溶剂thf和乙醚,剩余物用柱层析的方法进行分离纯化,以正己烷为展开剂。将过柱纯化得到的产物用氢化钙搅拌干燥5h后减压蒸馏,将减压蒸馏的产物加入到二丁基镁的庚烷溶液中搅拌3h后减压蒸馏,得到纯净的聚合单体vpdms。
第三步,合成si-h功能化聚合物主链pvpdms
在无水无氧、氩气保护的手套箱中,采用活性阴离子聚合的方法,以苯为溶剂,以sec-buli作为引发剂,引发单体vpdms的聚合反应。在聚合瓶中依次加入20ml苯,引发剂sec-buli(0.498mol·l-1,0.57ml)和vpdms单体(1g,0.0062mol),反应溶液迅速变成橙黄色。25℃下反应5h,加入精制的异丙醇终止反应。将反应溶液沉胶于400ml的甲醇中,抽滤,得到的固体粉末干燥至恒重,得到si-h功能化聚合主链pvpdms。聚合物数均分子量为3.5kg·mol-1,分子量分布指数为1.07。
第四步,合成含偶氮苯基侧链型液晶聚合物
在无水无氧、氩气保护下的手套箱中,将pvpdms3.5k(200mg,1.21mmolsi-h)加入到100ml安瓿瓶中。称取液晶单体m(0.58g,1.33mmolc=c)溶解至30ml无水无氧处理后的甲苯中,混合均匀后,加入到上述的安瓿瓶中,滴加4滴卡斯特催化剂(所用的卡斯特催化剂溶解于二甲苯溶液中,质量浓度为2%),引发硅氢加成反应。从手套箱中取出,60℃油浴中反应48h。反应结束后,减压旋蒸脱除溶剂,将反应物用少量的四氢呋喃溶解,向溶液中滴加乙醚有固体析出,抽滤,将得到的固体重复上述操作2-3次,得到纯净的侧链型液晶聚合物pvpdms3.5k-m。液晶聚合物数均分子量为13.8kg·mol-1,分子量分布指数为1.30。该液晶聚合物产生了微相分离,具有两个玻璃化转变温度,分别为聚合主链产生的玻璃化转变温度tg1为25℃,侧链液晶导致的tg2为77℃。
- 还没有人留言评论。精彩留言会获得点赞!